
N-Terminal Fragments of TDP-43—In Vitro Analysis and Implication in the Pathophysiology of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Constructions and Transfections
2.3. RT-PCR Analysis and Western Blot
2.4. Immunocytochemistry Analysis
2.5. Soluble and Insoluble Protein Fractions Analysis
2.6. Viability Assays
3. Results
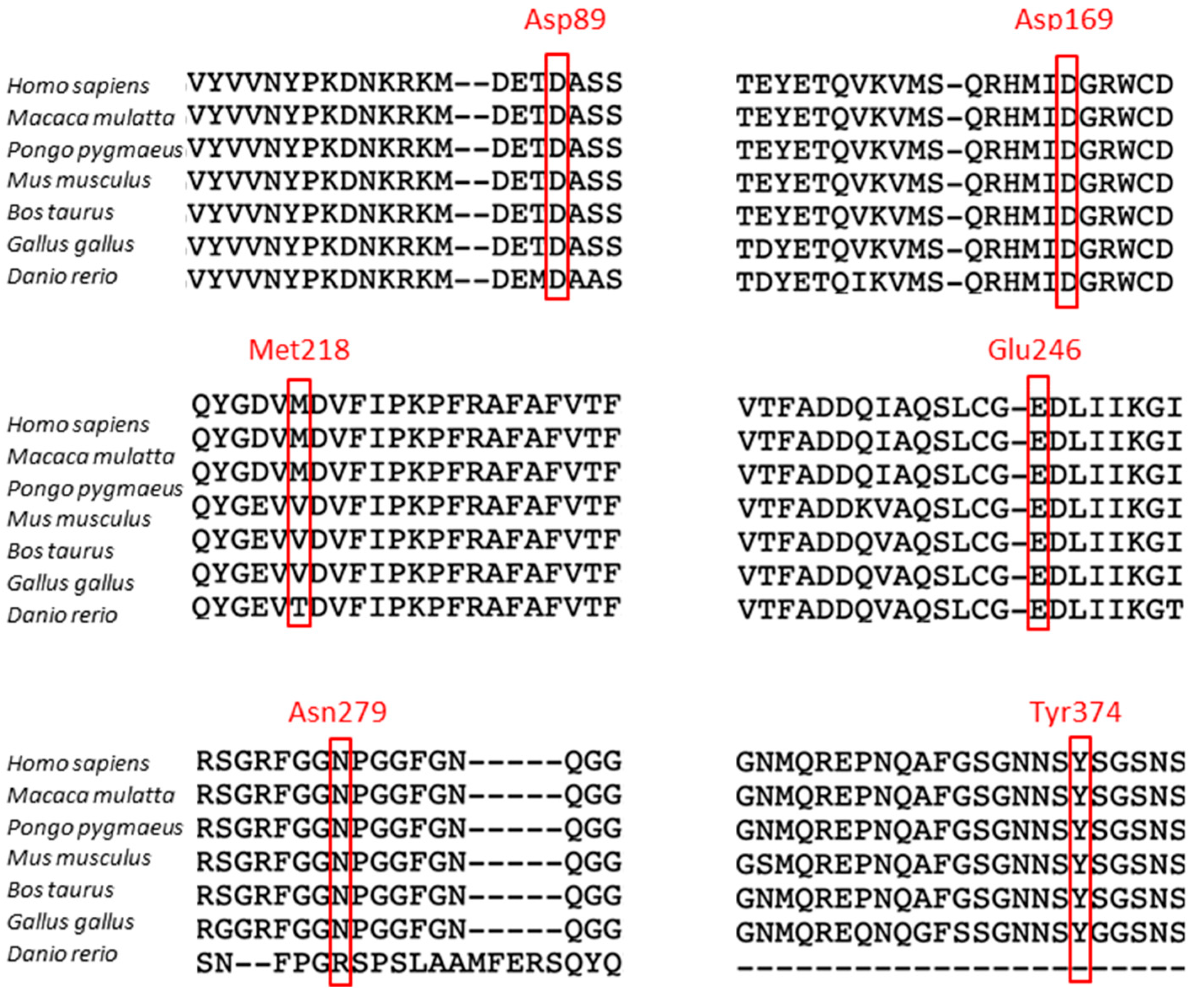
3.1. Cleavage Sites in the TDP-43 Protein
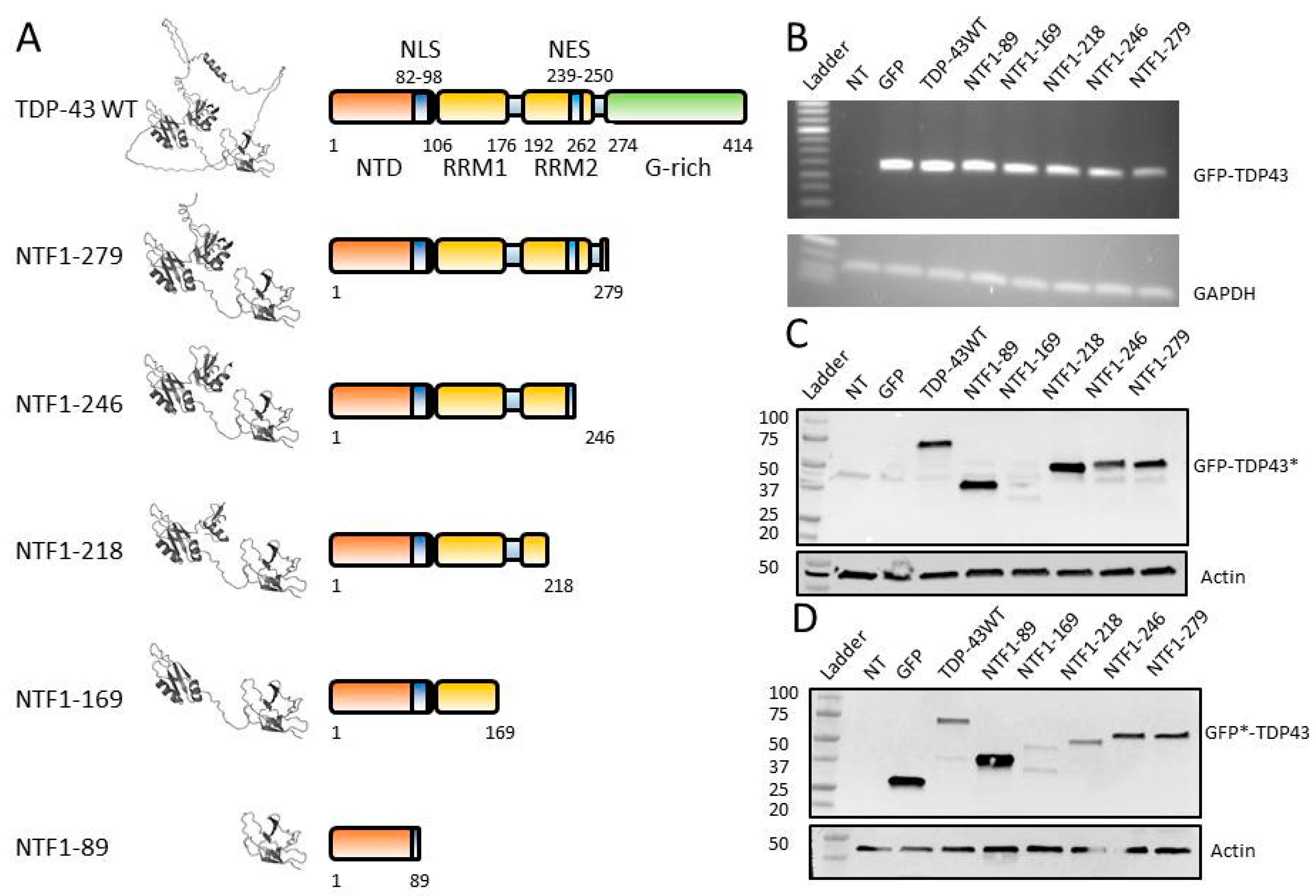
3.2. Construction of NTFs for In Vitro Studies
3.3. Expression of NTFs in HEK-293T
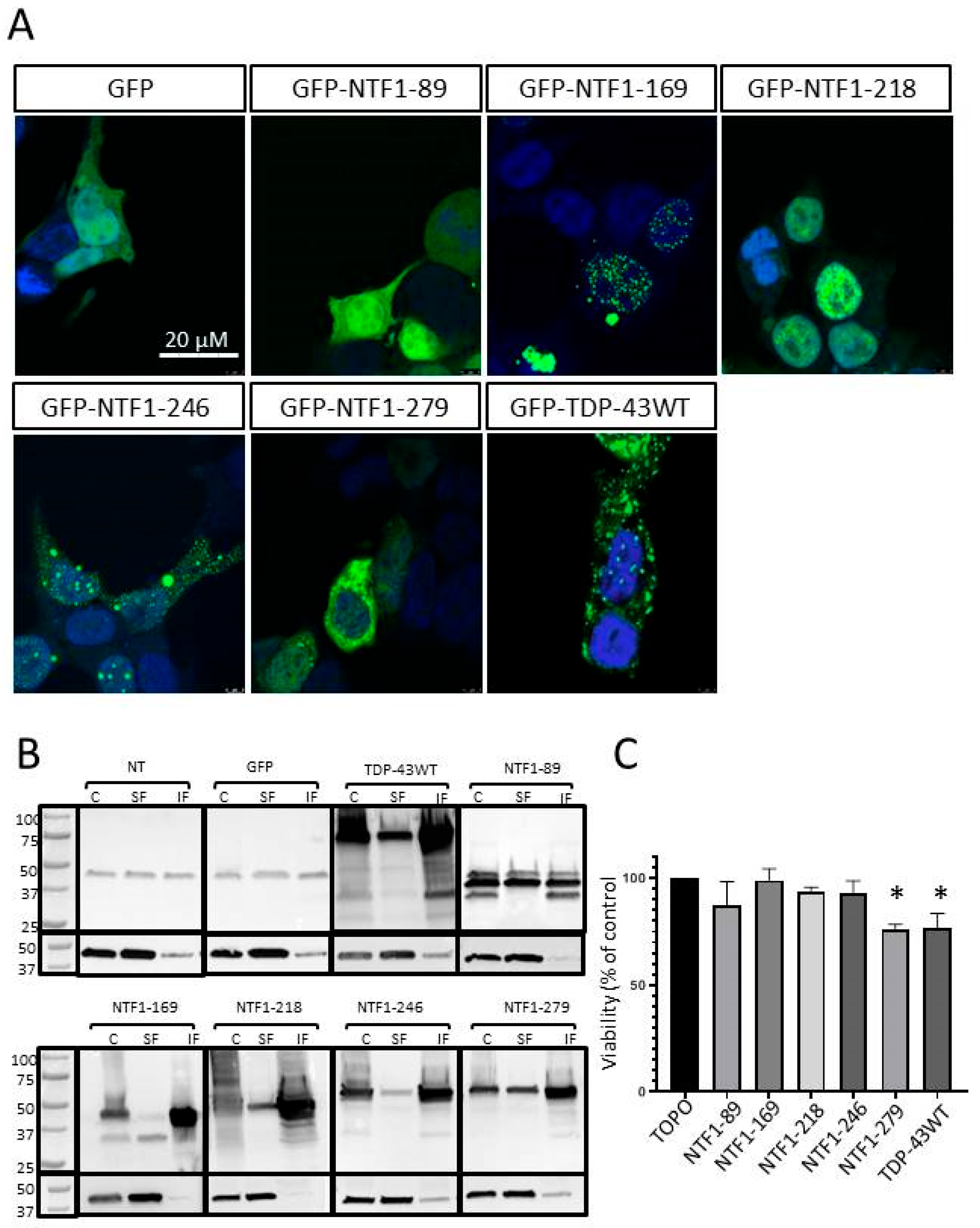
3.4. Effects of NTF Expression on Aggregate Formation
3.5. Effects of NTFs on Cell Viability
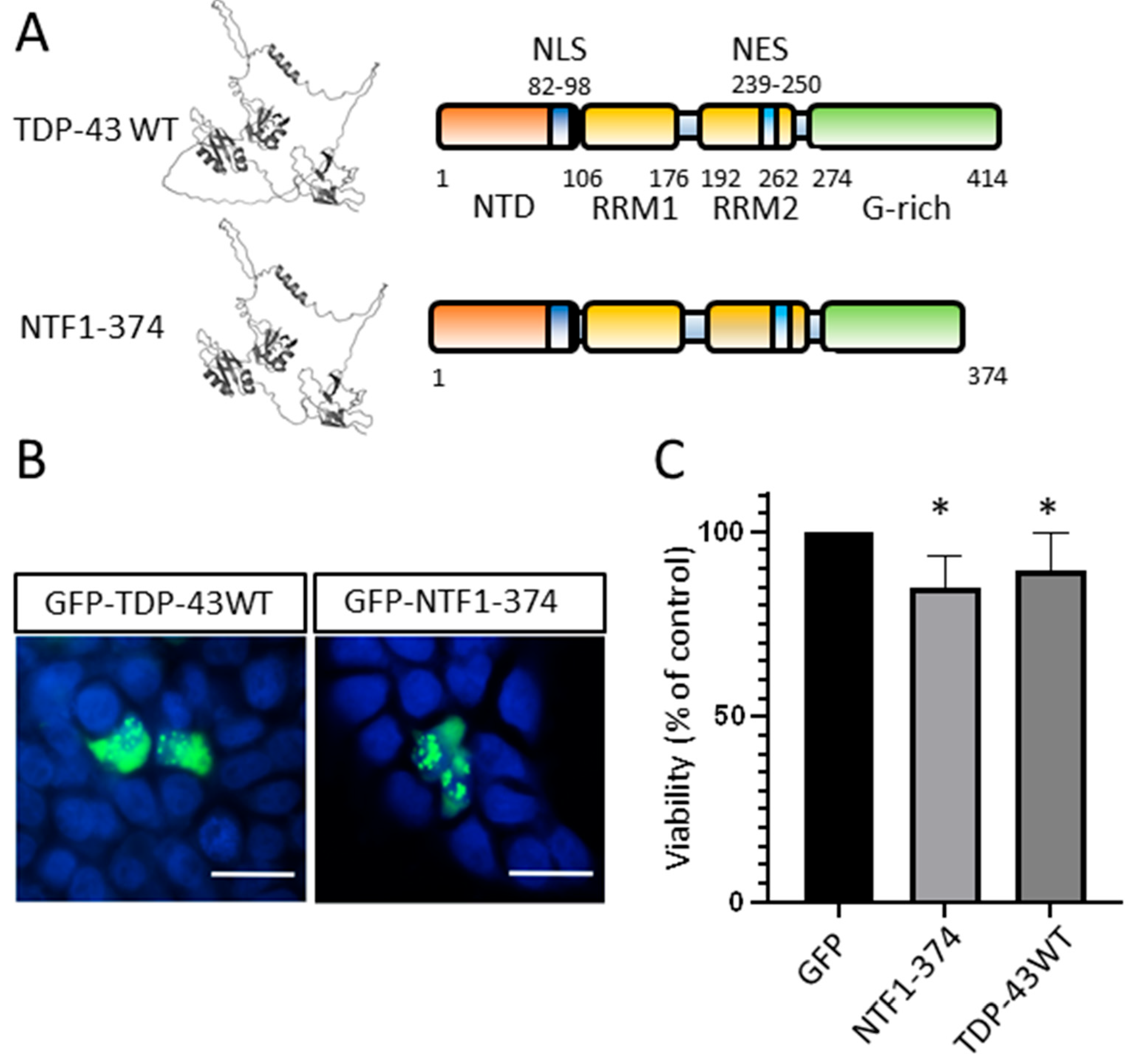
3.6. N-Terminal Fragment of TDP-43 Produced by Mutations in ALS Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ho, P.-C.; Hsieh, T.-C.; Tsai, K.-J. TDP-43 Proteinopathy in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis: From Pathomechanisms to Therapeutic Strategies. Ageing Res. Rev. 2024, 100, 102441. [Google Scholar] [CrossRef]
- Corcia, P.; Camu, W.; Brulard, C.; Marouillat, S.; Couratier, P.; Camdessanché, J.-P.; Cintas, P.; Verschueren, A.; Soriani, M.-H.; Desnuelle, C.; et al. Effect of Familial Clustering in the Genetic Screening of 235 French ALS Families. J. Neurol. Neurosurg. Psychiatry 2021, 92, 479–484. [Google Scholar] [CrossRef]
- Nijs, M.; Van Damme, P. The Genetics of Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 2024. [Google Scholar] [CrossRef]
- Mol, M.O.; van Rooij, J.G.J.; Wong, T.H.; Melhem, S.; Verkerk, A.J.M.H.; Kievit, A.J.A.; van Minkelen, R.; Rademakers, R.; Pottier, C.; Kaat, L.D.; et al. Underlying Genetic Variation in Familial Frontotemporal Dementia: Sequencing of 198 Patients. Neurobiol. Aging 2021, 97, 148.e9–148.e16. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 Regulates Its mRNA Levels through a Negative Feedback Loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Caulfield, T.; Xu, Y.-F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The Dual Functions of the Extreme N-Terminus of TDP-43 in Regulating Its Biological Activity and Inclusion Formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef]
- Sun, C.-S.; Wang, C.Y.-H.; Chen, B.P.-W.; He, R.-Y.; Liu, G.C.-H.; Wang, C.-H.; Chen, W.; Chern, Y.; Huang, J.J.-T. The Influence of Pathological Mutations and Proline Substitutions in TDP-43 Glycine-Rich Peptides on Its Amyloid Properties and Cellular Toxicity. PLoS ONE 2014, 9, e103644. [Google Scholar] [CrossRef]
- Maurel, C.; Madji-Hounoum, B.; Thepault, R.-A.; Marouillat, S.; Brulard, C.; Danel-Brunaud, V.; Camdessanche, J.-P.; Blasco, H.; Corcia, P.; Andres, C.R.; et al. Mutation in the RRM2 Domain of TDP-43 in Amyotrophic Lateral Sclerosis with Rapid Progression Associated with Ubiquitin Positive Aggregates in Cultured Motor Neurons. Amyotroph. Lateral Scler. Frontotemporal Degener. 2018, 19, 149–151. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 Proteinopathies: A New Wave of Neurodegenerative Diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 Control in Neurodegeneration: Autoregulation, Localization and Aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Lanznaster, D.; Hergesheimer, R.; Vourc’h, P.; Corcia, P.; Blasco, H. TDP43 Aggregates: The “Schrödinger’s Cat” in Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2021, 22, 514. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The Debated Toxic Role of Aggregated TDP-43 in Amyotrophic Lateral Sclerosis: A Resolution in Sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Cascella, R.; Capitini, C.; Fani, G.; Dobson, C.M.; Cecchi, C.; Chiti, F. Quantification of the Relative Contributions of Loss-of-Function and Gain-of-Function Mechanisms in TAR DNA-Binding Protein 43 (TDP-43) Proteinopathies. J. Biol. Chem. 2016, 291, 19437–19448. [Google Scholar] [CrossRef]
- Kitamura, A.; Nakayama, Y.; Shibasaki, A.; Taki, A.; Yuno, S.; Takeda, K.; Yahara, M.; Tanabe, N.; Kinjo, M. Interaction of RNA with a C-Terminal Fragment of the Amyotrophic Lateral Sclerosis-Associated TDP43 Reduces Cytotoxicity. Sci. Rep. 2016, 6, 19230. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates Its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef] [PubMed]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential Transformation of Human Neuronal Cells by Human Adenoviruses and the Origin of HEK 293 Cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Chami, A.A.; Beltran, S.; Corcia, P.; Andres, C.R.; Laumonnier, F.; Blasco, H.; Vourc’H, P. A Novel Mutation in the Cleavage Site N291 of TDP-43 Protein in a Familial Case of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Xu, Y.-F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin Mediates Caspase-Dependent Cleavage of TAR DNA Binding Protein-43. J. Neurosci. 2007, 27, 10530–10534. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Obi, T.; Shishido, T.; Akatsu, H.; Murayama, S.; Saito, Y.; Yoshida, M.; Hasegawa, M. Mass Spectrometric Analysis of Accumulated TDP-43 in Amyotrophic Lateral Sclerosis Brains. Sci. Rep. 2016, 6, 23281. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Lee, K.; Matsuoka, M. TDP-43-Induced Death Is Associated with Altered Regulation of BIM and Bcl-xL and Attenuated by Caspase-Mediated TDP-43 Cleavage. J. Biol. Chem. 2011, 286, 13171–13183. [Google Scholar] [CrossRef]
- Li, Q.; Yokoshi, M.; Okada, H.; Kawahara, Y. The Cleavage Pattern of TDP-43 Determines Its Rate of Clearance and Cytotoxicity. Nat. Commun. 2015, 6, 6183. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M.; et al. Enrichment of C-Terminal Fragments in TAR DNA-Binding Protein-43 Cytoplasmic Inclusions in Brain but Not in Spinal Cord of Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and Pathogenic Mutations Facilitate the Formation of Intracellular Aggregates of TDP-43. Human. Molecular Genetics 2009, 18, 3353–3364. [Google Scholar] [CrossRef]
- Yang, Z.; Lin, F.; Robertson, C.S.; Wang, K.K. Dual Vulnerability of TDP-43 to Calpain and Caspase-3 Proteolysis after Neurotoxic Conditions and Traumatic Brain Injury. J. Cereb. Blood Flow. Metab. 2014, 34, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.-Y. Expression of TDP-43 C-Terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 Loss of Function Increases TFEB Activity and Blocks Autophagosome-Lysosome Fusion. EMBO J. 2015, 35, 121–142. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A Role for Calpain-Dependent Cleavage of TDP-43 in Amyotrophic Lateral Sclerosis Pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial Loss of TDP-43 Function Causes Phenotypes of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Gozal, Y.M.; Duong, D.M.; Dammer, E.B.; Gearing, M.; Ye, K.; Lah, J.J.; Peng, J.; Levey, A.I.; Seyfried, N.T. Asparaginyl Endopeptidase Cleaves TDP-43 in Brain. Proteomics 2012, 12, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Furukawa, Y.; Suzuki, Y.; Fukuoka, M.; Nagasawa, K.; Nakagome, K.; Shimizu, H.; Mukaiyama, A.; Akiyama, S. A Molecular Mechanism Realizing Sequence-Specific Recognition of Nucleic Acids by TDP-43. Sci. Rep. 2016, 6, 20576. [Google Scholar] [CrossRef]
- Daoud, H.; Valdmanis, P.N.; Kabashi, E.; Dion, P.; Dupré, N.; Camu, W.; Meininger, V.; Rouleau, G.A. Contribution of TARDBP Mutations to Sporadic Amyotrophic Lateral Sclerosis. J. Med. Genet. 2009, 46, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A Comprehensive Analysis of Rare Genetic Variation in Amyotrophic Lateral Sclerosis in the UK. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Julian, T.H.; Feneberg, E.; Highley, J.R.; Sidra, M.; Turner, M.R.; Talbot, K.; Ansorge, O.; Allen, S.P.; Moll, T.; et al. Atypical TDP-43 Protein Expression in an ALS Pedigree Carrying a p.Y374X Truncation Mutation in TARDBP. Brain Pathol. 2023, 33, e13104. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and Dynamic Polymerization of the ALS-Linked Protein TDP-43 Antagonizes Its Pathologic Aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Xue, W.; Hong, J.-Y.; Zhang, J.-T.; Li, M.-J.; Yu, S.-N.; He, J.-H.; Hu, H.-Y. The N-Terminal Dimerization Is Required for TDP-43 Splicing Activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef]
- Chhangani, D.; Martín-Peña, A.; Rincon-Limas, D.E. Molecular, Functional, and Pathological Aspects of TDP-43 Fragmentation. iScience 2021, 24, 102459. [Google Scholar] [CrossRef]
- Sasaguri, H.; Chew, J.; Xu, Y.-F.; Gendron, T.F.; Garrett, A.; Lee, C.W.; Jansen-West, K.; Bauer, P.O.; Perkerson, E.A.; Tong, J.; et al. The Extreme N-Terminus of TDP-43 Mediates the Cytoplasmic Aggregation of TDP-43 and Associated Toxicity in Vivo. Brain Res. 2016, 1647, 57–64. [Google Scholar] [CrossRef]
- Pinarbasi, E.S.; Cağatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active Nuclear Import and Passive Nuclear Export Are the Primary Determinants of TDP-43 Localization. Sci. Rep. 2018, 8, 7083. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Disturbance of Nuclear and Cytoplasmic TAR DNA-Binding Protein (TDP-43) Induces Disease-like Redistribution, Sequestration, and Aggregate Formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| TDP-43WT | 5′-TAGTCTGAATATATTCGGGTAACCGAAG-3′ | 5′-GTTCGGCACTGGATATATGAACGCT-3′ |
| NTF1-89 | 5′-ATCTGTCTCATCCATTTTTC-3′ | 5-TAGTCATCAGCAGTGAAAGTG-3′ |
| NTF1-169 | 5-TAGTCATCAGCAGTGAAAGTG-3′ | 5′-TATGATAGATTAGCGATGGTGTGACTGC-3′ |
| NTF1-218 | 5-CCGTACTGAGAGAAGAAC-3′ | 5′-GGATGTGATGTAGGTCTTCATCC -3′ |
| NTF1-246 | 5′-AGAGACTGCGCAATCTGA -3′ | 5- TTGTGGAGAGTAGTTGATCATTAAAGGAATC-3′ |
| NTF1-279 | 5′-ATTACCACCAAATCTTCCAC-3′ | 5-TAGGGTGGCTTTGGGAATCAG-3′ |
| NTF1-374 | 5′-ATAACTCTTAGAGTGGCTCTAATTCTG-3′ | 5′TTCCAGAACCGAAGGCCT-3′ |
| Cleavage Sites | Fragments Size | Enzymes | References | Variants |
|---|---|---|---|---|
| Asp13-Glu14 | 42 kDa | Caspases 3, 7 | [25] | No |
| Arg55-Leu56 | 43–45 kDa | ND | [26] | No |
| Val75-Asn76 | 15–20 kDa | ND | [26] | No |
| Asp89-Ala90 | 35 kDa | Caspases 3, 7 | [25,27] | A90V |
| Val108-Leu109 | 30–35 kDa | ND | [26] | No |
| Val130-Leu131 | 30–35 kDa | ND | [26] | No |
| Asp169-Gly170 | 25 kDa | Caspase | [27,28] | D169G |
| Asp174-Cys175 | 25 kDa | Caspase 4 | [28] | No |
| Cys175-Lys176 | 23–25 kDa | ND | [26] | No |
| Leu207-Arg208 | <22 kDa | ND | [29,30] | No |
| Arg208-209 | 29 kDa | Caspase | [31] | No |
| Tyr214-Gly215 | 15–20 kDa | ND | [26] | No |
| Met218-Asp219 | 23 kDa | Caspases 3, 7 | [30,31,32] | No |
| Phe229-Ala230 | 25 kDa | Calpain | [31,33,34] | No |
| Leu243-Cys244 | 25 kDa | Calpain | [34,35] | No |
| Glu246-Asp247 | 25 kDa | Calpain | [30,32,35] | No |
| Asn279-Phe280 | 23 kDa | Calpain | [30] | No |
| Gln286-Gly287 | 34–37 kDa | Calpain | [31,33,34] | G287S |
| Asn291-Ser292 | 35 kDa | AEP | [26,36] | N291H |
| Gly295-Gly296 | 34–37 kDa | Calpain | [31,33,34] | G295C,R,S |
| Ala297-Gly298 | 34–37 kDa | Calpain | [31,33,34] | G298S |
| Asn-306-Met307 | 32 kDa | AEP | [36] | No |
| Met323-Ala324 | 37 à 39 kDa | Calpain | [31,33,34] | No |
| Phe401-Gly402 | ND | ND | [30] | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chami, A.A.; Bedja-Iacona, L.; Richard, E.; Lanznaster, D.; Marouillat, S.; Veyrat-Durebex, C.; Andres, C.R.; Corcia, P.; Blasco, H.; Vourc’h, P. N-Terminal Fragments of TDP-43—In Vitro Analysis and Implication in the Pathophysiology of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Genes 2024, 15, 1157. https://doi.org/10.3390/genes15091157
Chami AA, Bedja-Iacona L, Richard E, Lanznaster D, Marouillat S, Veyrat-Durebex C, Andres CR, Corcia P, Blasco H, Vourc’h P. N-Terminal Fragments of TDP-43—In Vitro Analysis and Implication in the Pathophysiology of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Genes. 2024; 15(9):1157. https://doi.org/10.3390/genes15091157
Chicago/Turabian StyleChami, Anna A., Léa Bedja-Iacona, Elodie Richard, Debora Lanznaster, Sylviane Marouillat, Charlotte Veyrat-Durebex, Christian R. Andres, Philippe Corcia, Hélène Blasco, and Patrick Vourc’h. 2024. "N-Terminal Fragments of TDP-43—In Vitro Analysis and Implication in the Pathophysiology of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration" Genes 15, no. 9: 1157. https://doi.org/10.3390/genes15091157