

A Novel Pathogenic Large Duplication in EXT1 Identified in a Family with Multiple Osteochondromas
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects of This Study
2.2. DNA Extraction
2.3. NGS Analysis
2.4. MLPA Analysis
2.5. qPCR Confirmation
2.6. LongPCR Amplification and Gel Electrophoresis
3. Results
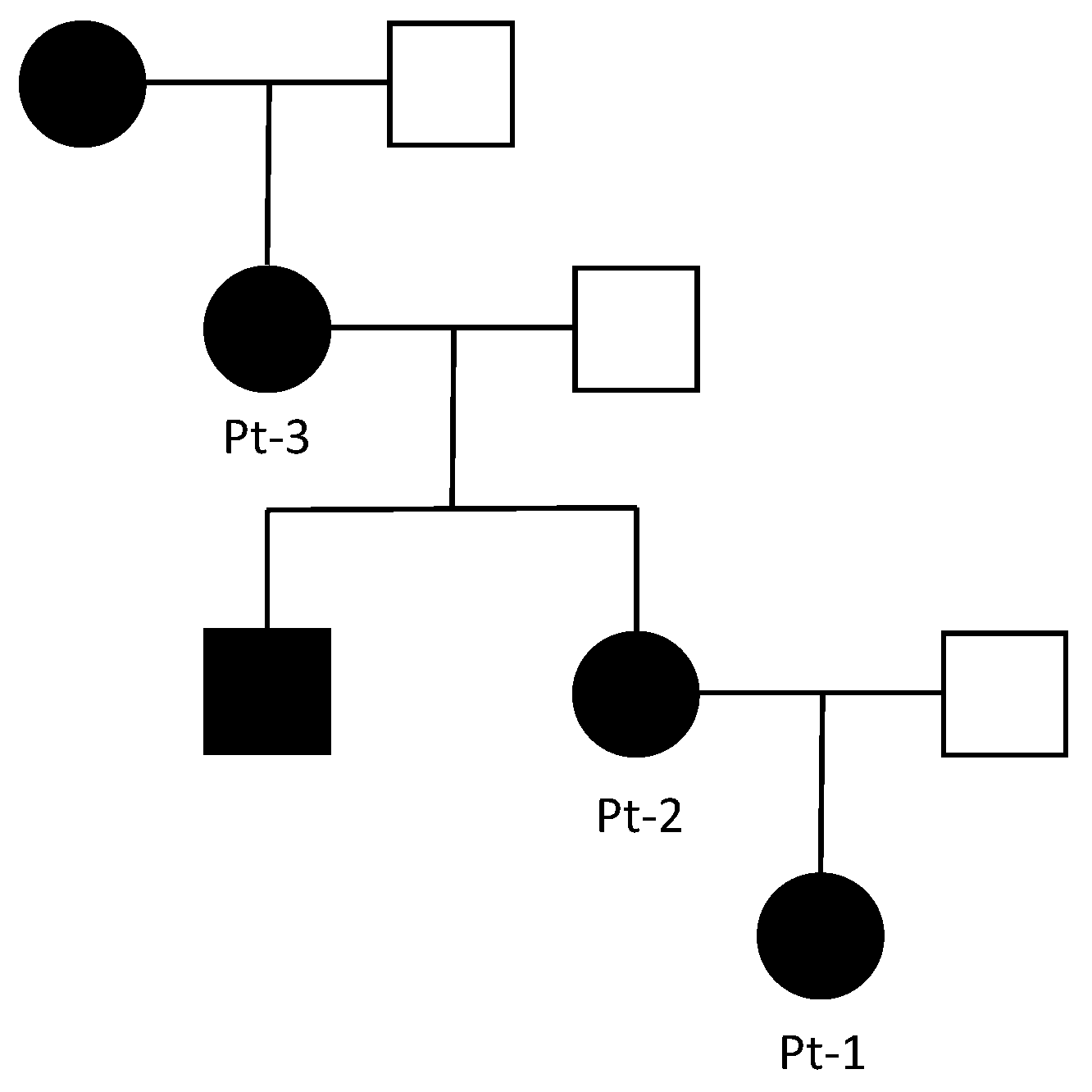
3.1. Clinical Report
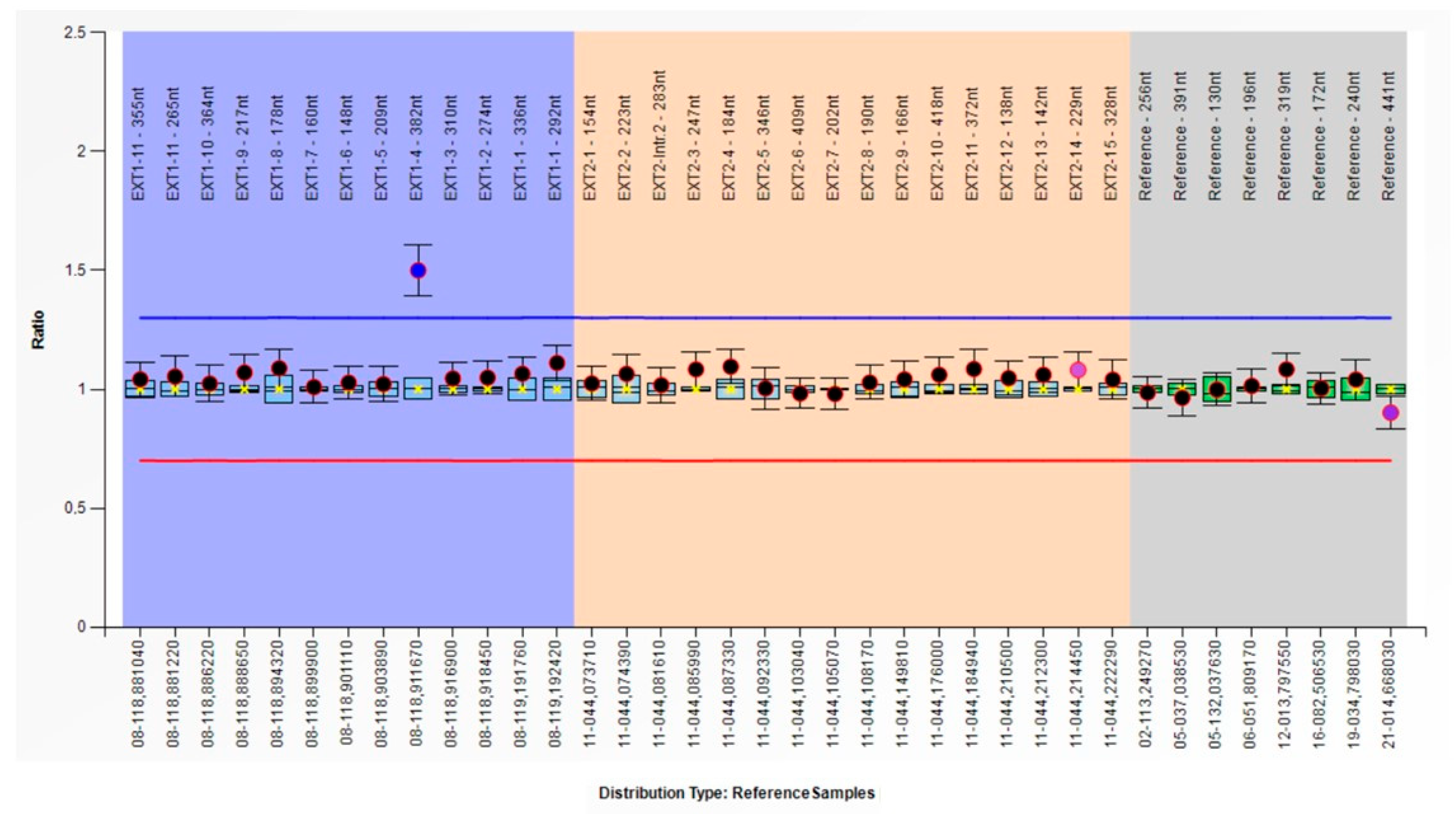
3.2. Genetic Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phan, A.Q.; Pacifici, M.; Esko, J.D. Advances in the pathogenesis and possible treatments for multiple hereditary exostoses from the 2016 international MHE conference. Connect. Tissue Res. 2018, 59, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Schmale, G.A.; Conrad, E.U.; Raskind, W.H. The natural history of hereditary multiple exostoses. J. Bone Jt. Surg. Am. 1994, 76, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Bovée, J.V.M.G. Multiple osteochondromas. Orphanet J. Rare Dis. 2008, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-Q.; Heutink, P.; de Vries, B.B.A.; Sandkuijl, L.A.; Ouweland, A.M.W.v.D.; Niermeijer, M.F.; Galjaard, H.; Reyniers, E.; Willems, P.J.; Halley, D.J. Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11. Hum. Mol. Genet. 1994, 3, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, W.W.E.W. Molecular basis of multiple exostoses: Mutations in the EXT1 and EXT2 genes. Hum. Mutat. 2000, 15, 220–227. [Google Scholar] [CrossRef]
- McCormick, C.; Duncan, G.; Goutsos, K.T.; Tufaro, F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. USA 2000, 97, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Huegel, J.; Sgariglia, F.; Enomoto-Iwamoto, M.; Koyama, E.; Dormans, J.P.; Pacifici, M. Heparan sulfate in skeletal development, growth, and pathology: The case of hereditary multiple exostoses. Dev. Dyn. 2013, 242, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Busse-Wicher, M.; Wicher, K.B.; Kusche-Gullberg, M. The exostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef]
- Bukowska-Olech, E.; Trzebiatowska, W.; Czech, W.; Drzymała, O.; Frąk, P.; Klarowski, F.; Kłusek, P.; Szwajkowska, A.; Jamsheer, A. Hereditary Multiple Exostoses—A Review of the Molecular Background, Diagnostics, and Potential Therapeutic Strategies. Front. Genet. 2021, 12, 759129. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Mordenti, M.; Gnoli, M.; Boarini, M.; Trisolino, G.; Evangelista, A.; Pedrini, E.; Corsini, S.; Tremosini, M.; Staals, E.L.; Antonioli, D.; et al. The Rizzoli Multiple Osteochondromas Classification revised: Describing the phenotype to improve clinical practice. Am. J. Med Genet. A 2021, 185, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Meregaglia, M.; Malandrini, F.; Finch, A.P.; Ciani, O.; Jommi, C. EQ-5D-5L Population Norms for Italy. Appl. Health Econ. Health Policy 2023, 21, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Boarini, M.; Tremosini, M.; Di Cecco, A.; Gnoli, M.; Brizola, E.; Mordenti, M.; Pedrini, E.; Locatelli, M.; Lanza, M.; Antonioli, D.; et al. Health-related quality of life and associated risk factors in patients with Multiple Osteochondromas: A cross-sectional study. Qual. Life Res. 2024, 33, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Sarrión, P.; Sangorrin, A.; Urreizti, R.; Delgado, A.; Artuch, R.; Martorell, L.; Armstrong, J.; Anton, J.; Torner, F.; Vilaseca, M.A.; et al. Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci. Rep. 2013, 3, 1346. [Google Scholar] [CrossRef]
- Porter, D.E.; Lonie, L.; Fraser, M.; Dobson-Stone, C.; Porter, J.R.; Monaco, A.P.; Simpson, A.H.R.W. Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study. J. Bone Jt. Surg. Br. 2004, 86, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, E.; Jennes, I.; Tremosini, M.; Milanesi, A.; Mordenti, M.; Parra, A.; Sgariglia, F.; Zuntini, M.; Campanacci, L.; Fabbri, N.; et al. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: Identification of “protective” and “risk” factors. J. Bone Jt. Surg. Am. 2011, 93, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Quenez, O.; Cassinari, K.; Coutant, S.; Lecoquierre, F.; Le Guennec, K.; Rousseau, S.; Richard, A.-C.; Vasseur, S.; Bouvignies, E.; Bou, J.; et al. Detection of copy-number variations from NGS data using read depth information: A diagnostic performance evaluation. Eur. J. Hum. Genet. 2021, 29, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Gabrielaite, M.; Torp, M.H.; Rasmussen, M.S.; Andreu-Sánchez, S.; Vieira, F.G.; Pedersen, C.B.; Kinalis, S.; Madsen, M.B.; Kodama, M.; Demircan, G.S.; et al. A Comparison of Tools for Copy-Number Variation Detection in Germline Whole Exome and Whole Genome Sequencing Data. Cancers 2021, 13, 6283. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLPA ex.3 | MLPA ex.4 | MLPA ex.5 | |
| Sample | Dosage Quotation (DQ) | ||
| Pt-1 | 1.05 | 1.5 | 1.02 |
| Pt-2 | 1.03 | 1.43 | 0.96 |
| Pt-3 | 1.01 | 1.54 | 1.05 |
| WT | 1.01 | 0.99 | 0.98 |
| qPCR ex.3 | qPCR ex.4 | qPCR ex.5 | |
| Sample | Relative Concentration (RC) | ||
| Pt-1 | 1.06 | 1.56 | 0.95 |
| Pt-2 | 1.04 | 1.51 | 0.99 |
| Pt-3 | 1.13 | 1.6 | 0.93 |
| WT | 1 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartolotti, I.; Sobul, K.; Corsini, S.; Scognamiglio, D.; Moroni, A.; Gnoli, M.; Sangiorgi, L.; Pedrini, E. A Novel Pathogenic Large Duplication in EXT1 Identified in a Family with Multiple Osteochondromas. Genes 2024, 15, 1169. https://doi.org/10.3390/genes15091169
Bartolotti I, Sobul K, Corsini S, Scognamiglio D, Moroni A, Gnoli M, Sangiorgi L, Pedrini E. A Novel Pathogenic Large Duplication in EXT1 Identified in a Family with Multiple Osteochondromas. Genes. 2024; 15(9):1169. https://doi.org/10.3390/genes15091169
Chicago/Turabian StyleBartolotti, Isabella, Klaudia Sobul, Serena Corsini, Davide Scognamiglio, Alice Moroni, Maria Gnoli, Luca Sangiorgi, and Elena Pedrini. 2024. "A Novel Pathogenic Large Duplication in EXT1 Identified in a Family with Multiple Osteochondromas" Genes 15, no. 9: 1169. https://doi.org/10.3390/genes15091169
APA StyleBartolotti, I., Sobul, K., Corsini, S., Scognamiglio, D., Moroni, A., Gnoli, M., Sangiorgi, L., & Pedrini, E. (2024). A Novel Pathogenic Large Duplication in EXT1 Identified in a Family with Multiple Osteochondromas. Genes, 15(9), 1169. https://doi.org/10.3390/genes15091169