
Multimodal Evaluation and Management of Wagner Syndrome—Three Patients from an Affected Family
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
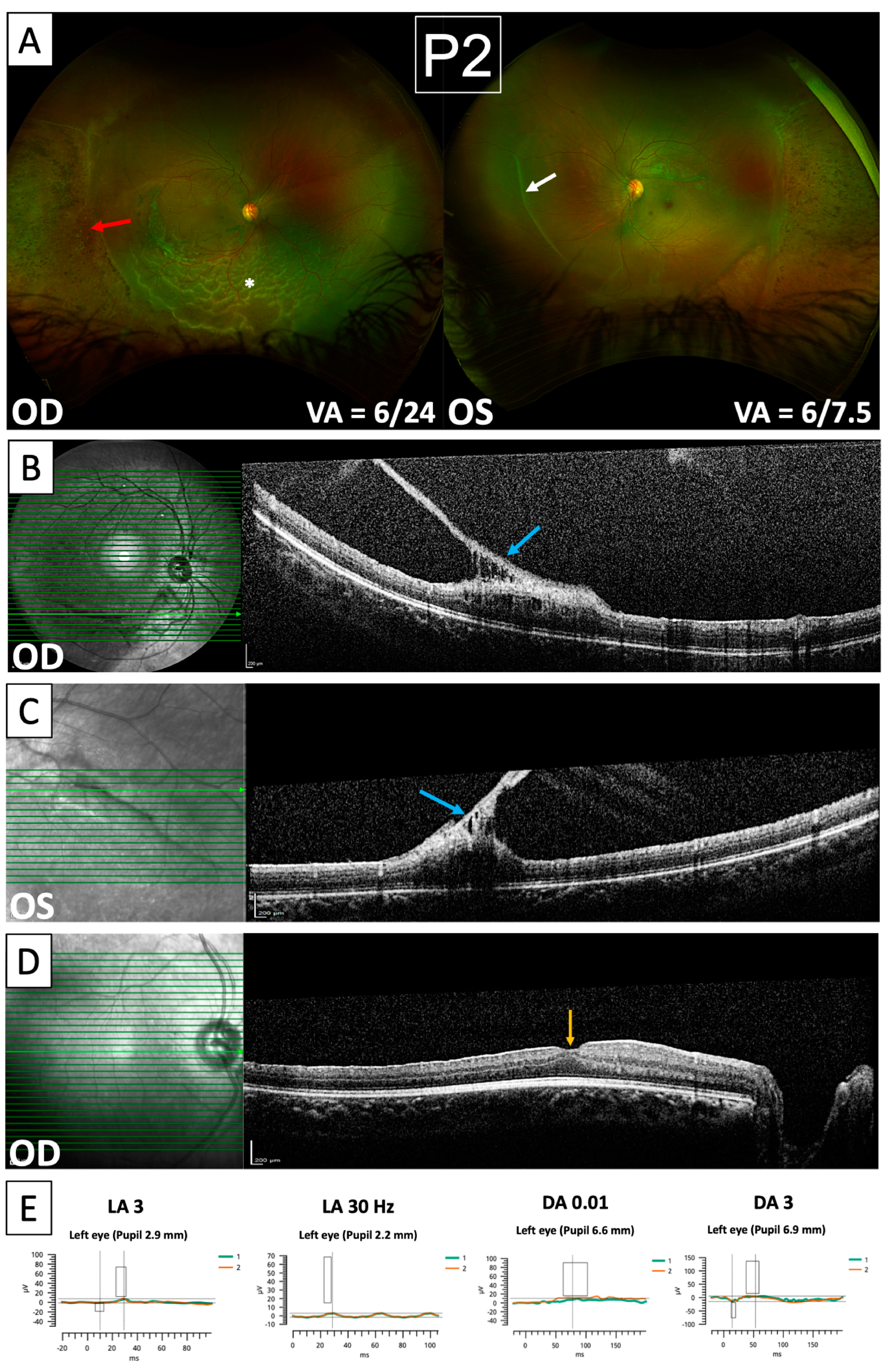
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Araújo, J.R.; Tavares-Ferreira, J.; Estrela-Silva, S.; Rocha, P.; Brandão, E.; Faria, P.A.; Falcão-Reis, F.; Rocha-Sousa, A. WAGNER syndrome: Anatomic, functional and genetic characterization of a Portuguese family. Graefe’s Arch. Clin. Exp. Ophthalmol. 2018, 256, 163–171. [Google Scholar] [CrossRef]
- Thomas, A.S.; Branham, K.; Van Gelder, R.N.; Daiger, S.P.; Sullivan, L.S.; Bowne, S.J.; Heckenlively, J.R.; Pennesi, M.E. Multimodal Imaging in Wagner Syndrome. Ophthalmic Surg. Lasers Imaging Retin. 2016, 47, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Inoue, H.; Sakamoto, Y.; Kudo, E.; Naito, T.; Mikawa, T.; Mikawa, Y.; Isashiki, Y.; Osabe, D.; Shinohara, S.; et al. Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Investig. Opthalmol. Vis. Sci. 2005, 46, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Nandadasa, S.; Foulcer, S.; Apte, S.S. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014, 35, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Watanabe, H. Versican: A Dynamic Regulator of the Extracellular Matrix. J. Histochem. Cyto-Chem. 2020, 68, 763–775. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Papageorgakopoulou, N.; Feretis, E.; Theocharis, D.A. Occurrence and structural characterization of versican-like proteoglycan in human vitreous. Biochimie 2002, 84, 1237–1243. [Google Scholar] [CrossRef]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Burns, T.A.; Dours-Zimmermann, M.T.; Zimmermann, D.R.; Krug, E.L.; Comte-Walters, S.; Reyes, L.; Davis, M.A.; Schey, K.L.; Schwacke, J.H.; Kern, C.B.; et al. Imbalanced expression of Vcan mRNA splice form proteins alters heart morphology and cellular protein profiles. PLoS ONE 2014, 9, e89133. [Google Scholar] [CrossRef]
- Graemiger, R.A.; Niemeyer, G.; Schneeberger, S.A.; Messmer, E.P. Wagner vitreoretinal degeneration. Follow-up of the original pedigree. Ophthalmology 1995, 102, 1830–1839. [Google Scholar] [CrossRef]
- Rothschild, P.-R.; Burin-Des-Roziers, C.; Audo, I.; Nedelec, B.; Valleix, S.; Brézin, A.P. Spectral-Domain Optical Coherence Tomography in Wagner Syndrome: Characterization of Vitreoretinal Interface and Foveal Changes. Arch. Ophthalmol. 2015, 160, 1065–1072.e1. [Google Scholar] [CrossRef] [PubMed]
- Borella, Y.; Dhaenens, C.-M.; Grunewald, O.; Caputo, G.; Borella, Y.; Dhaenens, C.-M.; Grunewald, O.; Caputo, G. Wagner syndrome: Novel VCAN variant and prophylactic management with encircling band and retinopexy. Am. J. Ophthalmol. Case Rep. 2024, 34, 102061. [Google Scholar] [CrossRef]
- Meredith, S.P.; Richards, A.J.; Flanagan, D.W.; Scott, J.D.; Poulson, A.V.; Snead, M.P. Clinical characterisation and molecular analysis of Wagner syndrome. Br. J. Ophthalmol. 2006, 91, 655–659. [Google Scholar] [CrossRef]
- Matsuyama, A.; Kalargyrou, A.A.; Smith, A.J.; Ali, R.R.; Pearson, R.A. A comprehensive atlas of Aggrecan, Versican, Neurocan and Phosphacan expression across time in wildtype retina and in retinal degeneration. Sci. Rep. 2022, 12, 7282. [Google Scholar] [CrossRef]
- Felemban, M.; Dorgau, B.; Hunt, N.C.; Hallam, D.; Zerti, D.; Bauer, R.; Ding, Y.; Collin, J.; Steel, D.; Krasnogor, N.; et al. Extracellular matrix component expression in human pluripotent stem cell-derived retinal organoids recapitulates retinogenesis in vivo and reveals an important role for IMPG1 and CD44 in the development of photoreceptors and interphotoreceptor matrix. Acta Biomater. 2018, 74, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.P.; Whitmore, S.S.; Lessing, N.D.; DeLuca, A.P.; Tucker, B.A.; Stone, E.M.; Mullins, R.F.; Scheetz, T.E. Spectacle: An interactive resource for ocular single-cell RNA sequencing data analysis. Exp. Eye Res. 2020, 200, 108204. [Google Scholar] [CrossRef]
- Dillinger, A.E.; Bauer, N.; Schneider, M.; Seitz, R.; Fuchshofer, R.; Tamm, E.R. Versican GAG-α domain deficiency causes rosette formation and detachment of the sensory retina in the mouse eye. Investig. Ophthalmol. Vis. Sci. 2022, 63, 1961-F0379. [Google Scholar]
- Ankala, A.; Jain, N.; Hubbard, B.; Alexander, J.J.; Shankar, S.P. Is exon 8 the most critical or the only dispensable exon of the VCAN gene? Insights into VCAN variants and clinical spectrum of Wagner syndrome. Am. J. Med. Genet. Part A 2018, 176, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Burin-Des-Roziers, C.; Rothschild, P.-R.; Layet, V.; Chen, J.-M.; Ghiotti, T.; Leroux, C.; Cremers, F.P.M.; Brézin, A.P.; Valleix, S. Deletions Overlapping VCAN Exon 8 Are New Molecular Defects for Wagner Disease. Hum. Mutat. 2017, 38, 43–47. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Nikopoulos, K.; Maugeri, A.; de Brouwer, A.P.M.; van Nouhuys, C.E.; Boon, C.J.F.; Perveen, R.; Zegers, H.A.A.; Wittebol-Post, D.; Biesen, P.R.v.D.; et al. Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Investig. Opthalmology Vis. Sci. 2006, 47, 3565–3572. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Bosserhoff, A.-K.; Nickl-Jockschat, T.; Doerfelt, A.; Bogdahn, U.; Hau, P. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-β2. Br. J. Cancer 2007, 96, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Wang, G.; Wang, Y.; Liang, J.; Wen, J.; Zheng, P.-S.; Wu, Y.; Lee, V.; Slingerland, J.; Dumont, D.; et al. The roles of versican V1 and V2 isoforms in cell proliferation and apoptosis. Mol. Biol. Cell 2005, 16, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Brézin, A.P.; Nedelec, B.; Barjol, A.; Rothschild, P.-R.; Delpech, M.; Valleix, S. A new VCAN/versican splice acceptor site mutation in a French Wagner family associated with vascular and inflammatory ocular features. Mol. Vis. 2011, 17, 1669–1678. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szeligowski, T.; Cehajic-Kapetanovic, J.; Raji, S.; Purohit, R.; Amin, H.; Patel, C.K.; Xue, K. Multimodal Evaluation and Management of Wagner Syndrome—Three Patients from an Affected Family. Genes 2024, 15, 1178. https://doi.org/10.3390/genes15091178
Szeligowski T, Cehajic-Kapetanovic J, Raji S, Purohit R, Amin H, Patel CK, Xue K. Multimodal Evaluation and Management of Wagner Syndrome—Three Patients from an Affected Family. Genes. 2024; 15(9):1178. https://doi.org/10.3390/genes15091178
Chicago/Turabian StyleSzeligowski, Tomasz, Jasmina Cehajic-Kapetanovic, Shabnam Raji, Ravi Purohit, Hoda Amin, Chetan K. Patel, and Kanmin Xue. 2024. "Multimodal Evaluation and Management of Wagner Syndrome—Three Patients from an Affected Family" Genes 15, no. 9: 1178. https://doi.org/10.3390/genes15091178