
A Novel MAG Variant Causes Hereditary Spastic Paraplegia in a Consanguineous Pakistani Family

 , , , , , ,
, , , , , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recruitment of Family
2.2. Genetic Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Panza, E.; Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: Genetic Heterogeneity and Common Pathways. Exp. Neurol. 2022, 357, 114203. [Google Scholar] [CrossRef] [PubMed]
- Bellofatto, M.; De Michele, G.; Iovino, A.; Filla, A.; Santorelli, F.M. Management of Hereditary Spastic Paraplegia: A Systematic Review of the Literature. Front. Neurol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Lossos, A.; Elazar, N.; Lerer, I.; Schueler-Furman, O.; Fellig, Y.; Glick, B.; Zimmerman, B.-E.; Azulay, H.; Dotan, S.; Goldberg, S.; et al. Myelin-Associated Glycoprotein Gene Mutation Causes Pelizaeus-Merzbacher Disease-like Disorder. Brain 2015, 138, 2521–2536. [Google Scholar] [CrossRef] [PubMed]
- Roda, R.H.; FitzGibbon, E.J.; Boucekkine, H.; Schindler, A.B.; Blackstone, C. Neurologic Syndrome Associated with Homozygous Mutation at MAG Sialic Acid Binding Site. Ann. Clin. Transl. Neurol. 2016, 3, 650–654. [Google Scholar] [CrossRef]
- McKerracher, L.; Rosen, K.M. MAG, Myelin and Overcoming Growth Inhibition in the CNS. Front. Mol. Neurosci. 2015, 8, 51. [Google Scholar] [CrossRef]
- Nguyen, T.; Mehta, N.R.; Conant, K.; Kim, K.J.; Jones, M.; Calabresi, P.A.; Melli, G.; Hoke, A.; Schnaar, R.L.; Ming, G.L.; et al. Axonal Protective Effects of the Myelin-Associated Glycoprotein. J. Neurosci. 2009, 29, 630–637. [Google Scholar] [CrossRef]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome Sequencing Links Corticospinal Motor Neuron Disease to Common Neurodegenerative Disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef]
- Roubertie, A.; Charif, M.; Meyer, P.; Manes, G.; Meunier, I.; Taieb, G.; Junta Morales, R.; Guichet, A.; Delettre, C.; Sarzi, E.; et al. Hereditary Spastic Paraplegia and Prominent Sensorial Involvement: Think MAG Mutations! Ann. Clin. Transl. Neurol. 2019, 6, 1572–1577. [Google Scholar] [CrossRef]
- Santos, M.; Damásio, J.; Kun-Rodrigues, C.; Barbot, C.; Sequeiros, J.; Brás, J.; Alonso, I.; Guerreiro, R. Novel MAG Variant Causes Cerebellar Ataxia with Oculomotor Apraxia: Molecular Basis and Expanded Clinical Phenotype. J. Clin. Med. 2020, 9, 1212. [Google Scholar] [CrossRef]
- Zech, M.; Brunet, T.; Škorvánek, M.; Blaschek, A.; Vill, K.; Hanker, B.; Hüning, I.; Haň, V.; Došekova, P.; Gdovinová, Z.; et al. Recessive Null-Allele Variants in MAG Associated with Spastic Ataxia, Nystagmus, Neuropathy, and Dystonia. Park. Relat. Disord. 2020, 77, 70–75. [Google Scholar] [CrossRef]
- Sager, G.; Turkyilmaz, A.; Ates, E.A.; Kutlubay, B. HACE1, GLRX5, and ELP2 Gene Variant Cause Spastic Paraplegies. Acta Neurol. Belg. 2022, 122, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Imran, R.; Naz, S. A Novel Homozygous Variant of SETX Causes Ataxia with Oculomotor Apraxia Type 2. J. Clin. Neurol. 2018, 14, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Vigeland, M.D.; Gjøtterud, K.S.; Selmer, K.K. FILTUS: A Desktop GUI for Fast and Efficient Detection of Disease-Causing Variants, Including a Novel Autozygosity Detector. Bioinformatics 2016, 32, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Saadi, S.M.; Cali, E.; Khalid, L.B.; Yousaf, H.; Zafar, G.; Khan, H.N.; Sher, M.; Vona, B.; Abdullah, U.; Malik, N.A.; et al. Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders. Genes 2023, 14, 1404. [Google Scholar] [CrossRef] [PubMed]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-Visual: A Free Online Tool to Improve SpliceAI Splicing Variant Interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef]
- Gleason, A.C.; Ghadge, G.; Chen, J.; Sonobe, Y.; Roos, R.P. Machine Learning Predicts Translation Initiation Sites in Neurologic Diseases with Nucleotide Repeat Expansions. PLoS ONE 2022, 17, e0256411. [Google Scholar] [CrossRef]
- Uctepe, E.; Vona, B.; Esen, F.N.; Sonmez, F.M.; Smol, T.; Tümer, S.; Mancılar, H.; Geylan Durgun, D.E.; Boute, O.; Moghbeli, M.; et al. Bi-Allelic Truncating Variants in CASP2 Underlie a Neurodevelopmental Disorder with Lissencephaly. Eur. J. Hum. Genet. 2024, 32, 52–60. [Google Scholar] [CrossRef]
- Tompson, S.W.; Young, T.L. Assaying the Effects of Splice Site Variants by Exon Trapping in a Mammalian Cell Line. Bio-Protocol 2017, 7, e2281. [Google Scholar] [CrossRef]
- Efthymiou, S.; Scala, M.; Nagaraj, V.; Ochenkowska, K.; Komdeur, F.L.; Liang, R.A.; Abdel-Hamid, M.S.; Sultan, T.; Barøy, T.; Van Ghelue, M.; et al. Novel Loss-of-Function Variants Expand ABCC9-Related Intellectual Disability and Myopathy Syndrome. Brain 2024, 147, 1822–1836. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Luo, W.J.; Yu, H.; Wang, P.S.; Dong, H.L.; Li, H.F.; Wu, Z.Y. A Novel PCYT2 Mutation Identified in a Chinese Consanguineous Family with Hereditary Spastic Paraplegia. J. Genet. Genom. 2021, 48, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; He, J.; Liu, X.; Wu, J.; Cai, X.; Zhang, Y.; Liu, X.; Fan, D. Clinical Features and Genetic Spectrum of Chinese Patients with Hereditary Spastic Paraplegia. Front. Genet. 2023, 14, 1085442. [Google Scholar] [CrossRef] [PubMed]
- Bereznyakova, O.; Dupré, N. Spastic Ataxias. Handb. Clin. Neurol. 2018, 155, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Schüle, R. Overcoming the Divide between Ataxias and Spastic Paraplegias: Shared Phenotypes, Genes, and Pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef]
- Pfeffer, G.; Pyle, A.; Griffin, H.; Miller, J.; Wilson, V.; Turnbull, L.; Fawcett, K.; Sims, D.; Eglon, G.; Hadjivassiliou, M.; et al. SPG7 Mutations Are a Common Cause of Undiagnosed Ataxia. Neurology 2015, 84, 1174–1177. [Google Scholar] [CrossRef]
- Van De Warrenburg, B.P.; Schouten, M.I.; De Bot, S.T.; Vermeer, S.; Meijer, R.; Pennings, M.; Gilissen, C.; Willemsen, M.A.; Scheffer, H.; Kamsteeg, E.J. Clinical Exome Sequencing for Cerebellar Ataxia and Spastic Paraplegia Uncovers Novel Gene-Disease Associations and Unanticipated Rare Disorders. Eur. J. Hum. Genet. 2016, 24, 1460–1466. [Google Scholar] [CrossRef]
- Georgiou, J.; Tropak, M.B.; Roder, J.C. Myelin-Associated Glycoprotein Gene. Myelin Biol. Disord. 2004, 1, 421–467. [Google Scholar] [CrossRef]
- Liu, B.P.; Fournier, A.; GrandPré, T.; Strittmatter, S.M. Myelin-Associated Glycoprotein as a Functional Ligand for the Nogo-66 Receptor. Science 2002, 297, 1190–1193. [Google Scholar] [CrossRef]
- Lopez, P.H.H. Role of Myelin-Associated Glycoprotein (Siglec-4a) in the Nervous System. Adv. Neurobiol. 2014, 9, 245–262. [Google Scholar] [CrossRef]
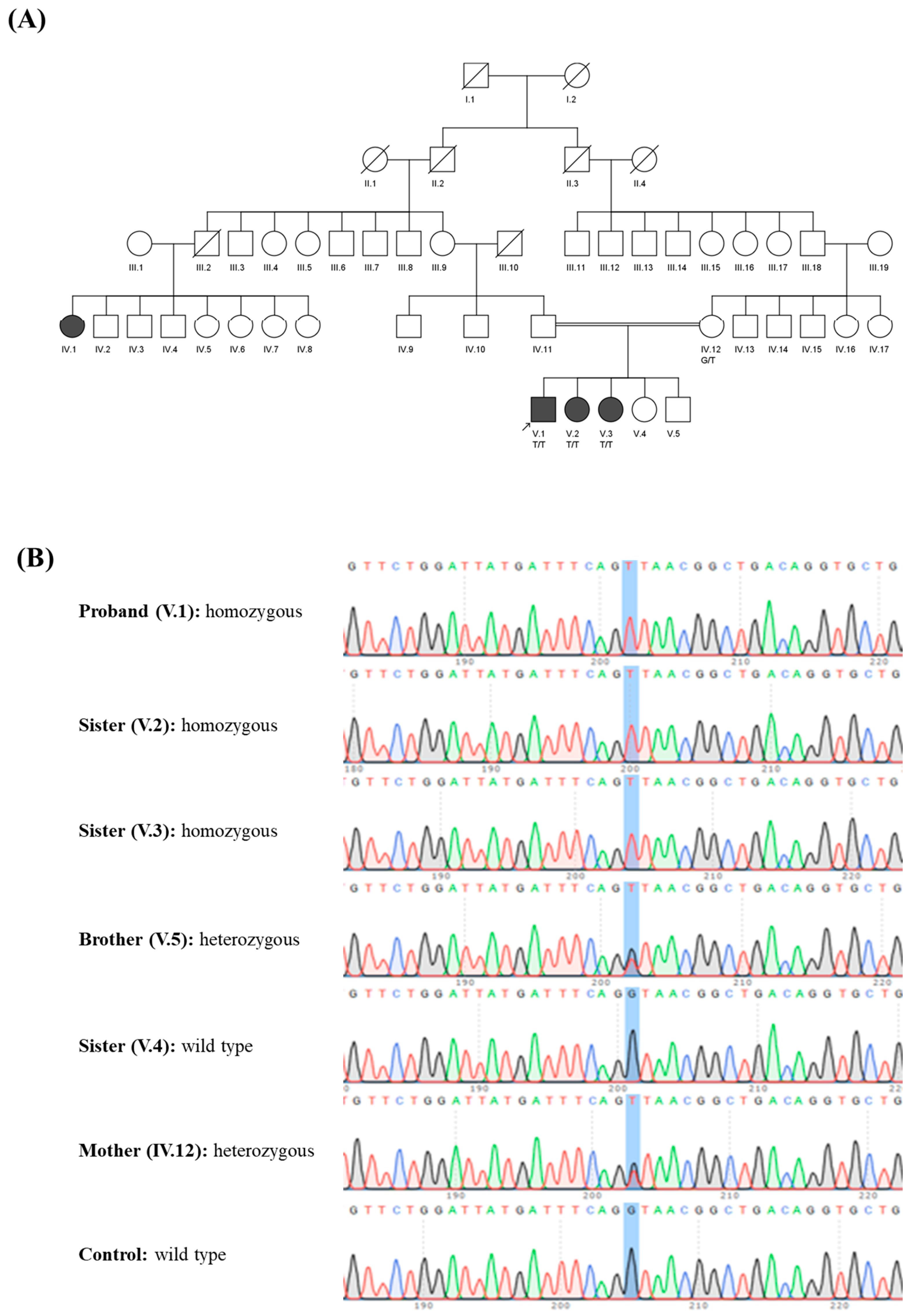

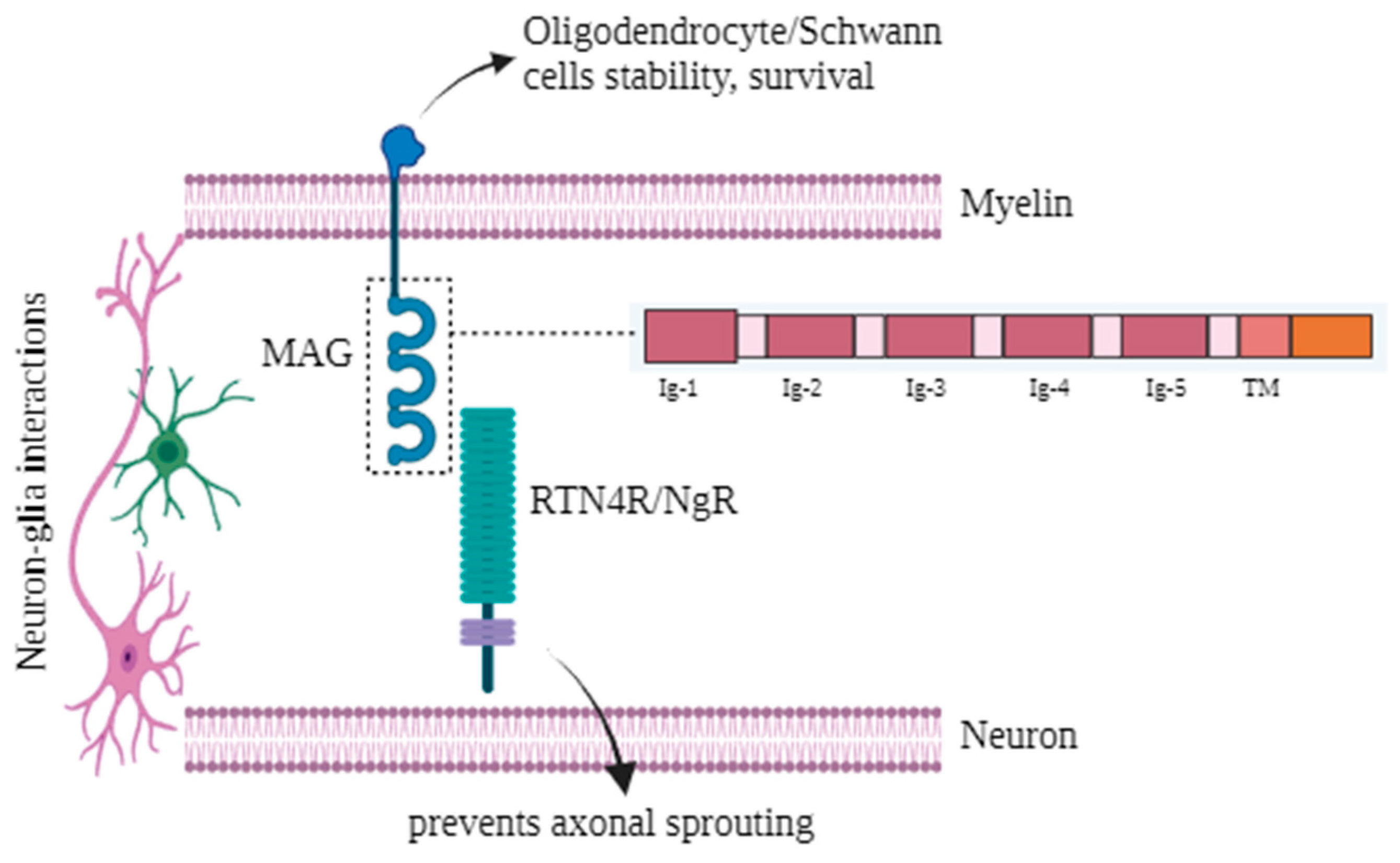

{kind=link}
{kind=link}
{kind=link}
| Present Study | Previous Studies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Identifier | V.1 | V.2 | V.3 | [7] 1226-IV-3 | [7] 1226-IV-4 | [3] III-2 | [3] III-3 | [3] III-5 | [4] II.2 | [4] II.5 | [8] |
| Transcript | NM_080600.3 | NM_080600.3 | NM_080600.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 |
| Mutation | c.46 + 1G > T | c.46 + 1G > T | c.46 + 1G > T | c.1288T > G (p.Cys430Gly) | c.1288T > G (p.Cys430Gly) | c.399C > G (p.Ser133Arg) | c.399C > G (p.Ser133Arg) | c.399C > G (p.Ser133Arg) | c.353G > A (p.Arg118His) | c.353G > A (p.Arg118His) | c.452C > T (p.Ala151Val); c.1117A > C (p.Ser373Arg) |
| Zygosity | Hom | Hom | Hom | Hom | Hom | Hom | Hom | Hom | Hom | Hom | Comp Het |
| Consequence | Splice donor | Splice donor | Splice donor | Missense | Missense | Missense | Missense | Missense | Missense | Missense | - |
| Sex | M | F | F | F | F | M | M | F | M | M | F |
| Age of onset (yr) | 1 | 1 | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Age of last examination (yr) | 17 | 15 | 13 | 12 | 11 | 28 | 26 | 21 | 54 | 48 | 10 |
| Ethnic background | Pakistan | Pakistan | Pakistan | N/A | N/A | Middle Eastern | Middle Eastern (Palestinian Arab) | Middle Eastern | N/A | N/A | African |
| Consanguinity | + | + | + | + | + | + | + | + | + | + | - |
| Developmental delay | + | + | + | N/A | N/A | + | + | + | + | N/A | + |
| Hypotonia | N/A | N/A | N/A | N/A | N/A | + | + | + | N/A | N/A | N/A |
| Intellectual disability | + | + | + | N/A | N/A | + | + | + | + | + | + |
| Cognitive impairment | + | + | + | + | + | + | + | + | + | + | + |
| Dysarthria | + | + | + | - | - | + | + | + | + | N/A | N/A |
| Nystagmus | + | + | + | + | + | + | + | + | + | + | N/A |
| Other ophthalmologic findings | + | + | + | - | - | + | + | + | + | + | + |
| Spasticity | + | + | + | + | + | + | + | + | + | - | + |
| Pyramidal signs | + | + | + | + | + | + | + | + | + | + | + |
| Ataxic signs | + | + | + | + | + | + | + | + | + | + | + |
| Dystonia | N/A | N/A | N/A | - | - | - | - | - | - | - | - |
| Hypo-/areflexia | + | + | + | - | - | + | + | + | + | + | + |
| Vibratory sense deficit | - | - | - | + | + | + | + | + | + | + | N/A |
| Muscular atrophy | N/A | N/A | N/A | + | + | + | + | + | N/A | N/A | + |
| Delay of gait | NA (Quadrupedal gait) | + | + | N/A | N/A | + | + | + | - | + | - |
| Neuropathy | N/A | N/A | N/A | N/A | N/A | + | + | + | + | + | + |
| Support for walking | N/A | - | - | N/A | N/A | + | + | + | N/A | N/A | + |
| Motor Deficit | + | + | + | + | + | + | + | + | + | + | + |
| Scoliosis | - | - | - | N/A | N/A | - | - | - | N/A | N/A | N/A |
| limb dysmetria | + | + | + | N/A | N/A | + | + | + | + | + | + |
| Intention tremor | + | + | + | N/A | N/A | - | - | - | N/A | N/A | N/A |
| Patient Identifier | [10] | [10] | [10] | [10] | [9] | [9] | [9] | [11] | [11] | ||
| A-II-1 | B-II-1 | C-II-1 | D-II-1 | II.1 | II.2 | II.3 | P13 | P14 | |||
| Transcript | NM_002361.3 | NM_002361.3 | NM_002361.3 | NM_002361.3 | - | - | NP_002352.1 | NM_002361 | NM_002361 | ||
| Mutation | c.1126C > T (p.Gln376) | c.517_521dup (p.Trp174) | c.1522C > T (p.Arg508) | c.693C > A (p.Tyr231); c.980G > A (p.Trp327) | c.124T > C(p.Cys42Arg) | c.124T > C (p.Cys42Arg) | c.124T > C(p.Cys42Arg) | c.475T > G (p.Cys159Gly) | c.475 T > G (p.Cys159Gly) | ||
| Zygosity | Hom | Hom | Hom | Comp Het | Hom | Hom | Hom | Hom | Hom | ||
| Consequence | Nonsense | Frameshift | Nonsense | Nonsense | Missense | Missense | Missense | N/A | N/A | ||
| Sex | M | F | F | F | M | M | F | M | F | ||
| Age of onset (yr) | Early infancy | N/A | N/A | N/A | 1 | 1 | 1 | 1-2 month | 1-2 month | ||
| Age of last examination (yr) | 35 | 21 | 2.5 | 12 | 59 | 56 | 54 | 10 | 3 | ||
| Ethnic background | European | European | Middle eastern | European | Portuguese | Portuguese | Portuguese | Turkish | Turkish | ||
| Consanguinity | - | + | + | - | + | + | + | + | + | ||
| Developmental delay | + | + | + | + | N/A | N/A | N/A | - | - | ||
| Hypotonia | + | - | + | + | N/A | N/A | N/A | + | + | ||
| Intellectual disability | + | + | + | + | N/A | N/A | N/A | - | - | ||
| Cognitive impairment | - | + | - | - | - | - | - | N/A | N/A | ||
| Dysarthria | + | + | - | + | N/A | N/A | N/A | + | + | ||
| Nystagmus | + | + | + | + | N/A | N/A | N/A | + | + | ||
| Other ophthalmologic findings | + | - | - | + | + (oculomotor apraxia) | ++ | ++ | - | - | ||
| Spasticity | + | + | - | + | - | - | - | + (in lower limbs only) | + (in lower limbs only) | ||
| Pyramidal signs | + | + | - | - | - | - | - | N/A | N/A | ||
| Ataxic signs | + | + | + | + | + (First sign) | + (First sign) | + (First sign) | ++ (12 month) | ++ (12 month) | ||
| Dystonia | + | + | - | - | - | - | - | ||||
| Hypo-/areflexia | + | - | + | + | - | - | - | N/A | N/A | ||
| Vibratory sense deficit | + | - | - | + | N/A | N/A | N/A | N/A | N/A | ||
| Muscular atrophy | + | - | - | - | N/A | N/A | N/A | N/A | N/A | ||
| Delay of gait | N/A | N/A | Wide base gait | N/A | + | + | + | N/A | N/A | ||
| Neuropathy | + | - | N/A | - | + (Prominent) | + (Prominent) | + (Prominent) | N/A | N/A | ||
| Support for walking | N/A | + | N/A | + | + | N/A | N/A | ||||
| Motor Deficit | + | + | + | + | +++ | +++ | +++ | - | - | ||
| Scoliosis | + | N/A | N/A | N/A | - | - | - | N/A | N/A | ||
| limb dysmetria | N/A | N/A | N/A | N/A | + | + | + | N/A | N/A | ||
| Intention tremor | N/A | N/A | N/A | N/A | N/A | + | + | N/A | N/A | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akram, R.; Anwar, H.; Muzaffar, H.; Turchetti, V.; Lau, T.; Vona, B.; Makhdoom, E.U.H.; Iqbal, J.; Mahmood Baig, S.; Hussain, G.; et al. A Novel MAG Variant Causes Hereditary Spastic Paraplegia in a Consanguineous Pakistani Family. Genes 2024, 15, 1203. https://doi.org/10.3390/genes15091203
Akram R, Anwar H, Muzaffar H, Turchetti V, Lau T, Vona B, Makhdoom EUH, Iqbal J, Mahmood Baig S, Hussain G, et al. A Novel MAG Variant Causes Hereditary Spastic Paraplegia in a Consanguineous Pakistani Family. Genes. 2024; 15(9):1203. https://doi.org/10.3390/genes15091203
Chicago/Turabian StyleAkram, Rabia, Haseeb Anwar, Humaira Muzaffar, Valentina Turchetti, Tracy Lau, Barbara Vona, Ehtisham Ul Haq Makhdoom, Javed Iqbal, Shahid Mahmood Baig, Ghulam Hussain, and et al. 2024. "A Novel MAG Variant Causes Hereditary Spastic Paraplegia in a Consanguineous Pakistani Family" Genes 15, no. 9: 1203. https://doi.org/10.3390/genes15091203