
Comparison of B-Cell Lupus and Lymphoma Using a Novel Immune Imbalance Transcriptomics Algorithm Reveals Potential Therapeutic Targets

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preprocessing of RNA-Sequencing Data
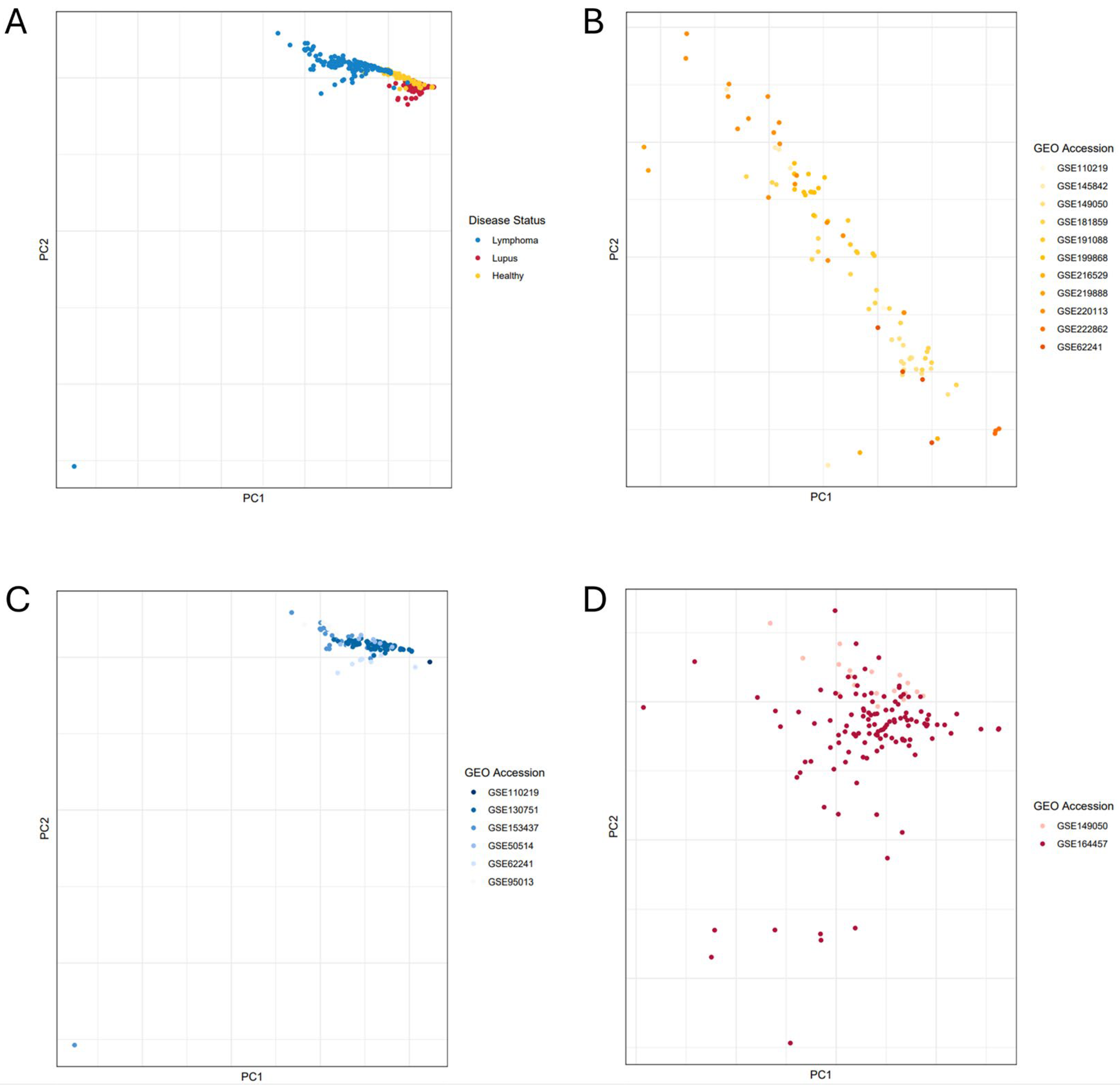
2.2. Principal Component Analysis
2.3. Immune Imbalance Determination
2.4. Immune Imbalance Validation
3. Results
3.1. IIT Algorithm Design Solutions
3.2. Lupus vs. Lymphoma B-Cell Comparison Using IIT Algorithm
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, J.; Zhang, D.; Yao, X.; Huang, Y.; Lu, Q. Global Epidemiology of Systemic Lupus Erythematosus: A Comprehensive Systematic Analysis and Modelling Study. Ann. Rheum. Dis. 2023, 82, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Furie, R. Novel Paradigms in Systemic Lupus Erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Gershwin, M.E. The Consequences of Apoptosis in Autoimmunity. J. Autoimmun. 2008, 31, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, P.; Cesareo, M.; Chimenti, M.S.; Triggianese, P.; Canofari, C.; Barbato, C.; Giannini, C.; Salandri, A.G.; Nucci, C.; Perricone, R. Take a Look at the Eyes in Systemic Lupus Erythematosus: A Novel Point of View. Autoimmun. Rev. 2019, 18, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Zonana-Nacach, A.; Barr, S.G.; Magder, L.S.; Petri, M. Damage in Systemic Lupus Erythematosus and Its Association with Corticosteroids. Arthritis Rheum. 2000, 43, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Urowitz, M.B.; Rahman, P.; Ibañez, D.; Tam, L.-S. Accrual of Organ Damage over Time in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2003, 30, 1955–1959. [Google Scholar] [PubMed]
- Basta, F.; Fasola, F.; Triantafyllias, K.; Schwarting, A. Systemic Lupus Erythematosus (SLE) Therapy: The Old and the New. Rheumatol. Ther. 2020, 7, 433–446. [Google Scholar] [CrossRef]
- Jamil, A.; Mukkamalla, S.K.R. Lymphoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef]
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin Lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile Strategy for Controlling the Specificity and Activity of Engineered T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The Risk of Lymphoma Development in Autoimmune Diseases: A Meta-Analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Headland, S.E.; Norling, L.V. The Resolution of Inflammation: Principles and Challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Rogovskii, V. Immune Tolerance as the Physiologic Counterpart of Chronic Inflammation. Front. Immunol. 2020, 11, 2061. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The Crucial Roles of Inflammatory Mediators in Inflammation: A Review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N.; Stockinger, B.; Tak, P.P. The Resolution of Inflammation. Nat. Rev. Immunol. 2013, 13, 59–66. [Google Scholar] [CrossRef]
- Delogu, L.G.; Deidda, S.; Delitala, G.; Manetti, R. Infectious Diseases and Autoimmunity. J. Infect. Dev. Ctries. 2011, 5, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kim-Hellmuth, S.; Bechheim, M.; Pütz, B.; Mohammadi, P.; Nédélec, Y.; Giangreco, N.; Becker, J.; Kaiser, V.; Fricker, N.; Beier, E.; et al. Genetic Regulatory Effects Modified by Immune Activation Contribute to Autoimmune Disease Associations. Nat. Commun. 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer Immunosuppression and Autoimmune Disease: Beyond Immunosuppressive Networks for Tumour Immunity. Immunology 2006, 119, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ghorani, E.; Swanton, C.; Quezada, S.A. Cancer Cell-Intrinsic Mechanisms Driving Acquired Immune Tolerance. Immunity 2023, 56, 2270–2295. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Menter, T.; Tzankov, A. Mechanisms of Immune Evasion and Immune Modulation by Lymphoma Cells. Front. Oncol. 2018, 8, 54. [Google Scholar] [CrossRef]
- Pardoll, D.M. Inducing Autoimmune Disease to Treat Cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 5340–5342. [Google Scholar] [CrossRef] [PubMed]
- Pennell, C.A.; Barnum, J.L.; McDonald-Hyman, C.S.; Panoskaltsis-Mortari, A.; Riddle, M.J.; Xiong, Z.; Loschi, M.; Thangavelu, G.; Campbell, H.M.; Storlie, M.D.; et al. Human CD19-Targeted Mouse T Cells Induce B Cell Aplasia and Toxicity in Human CD19 Transgenic Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1423–1434. [Google Scholar] [CrossRef]
- Adil, S.; Paracha, R.Z.; Tariq, S.; Nisar, M.; Ijaz, S.; Siddiqa, A.; Hussain, Z.; Amir, A. A Computational Systems Analyses to Identify Biomarkers and Mechanistic Link in Psoriasis and Cutaneous Squamous Cell Carcinoma. Front. Immunol. 2021, 12, 662528. [Google Scholar] [CrossRef]
- Seligman, C.; Chang, Y.-M.; Luo, J.; Garden, O.A. Exploring the Role of Immune Checkpoint Inhibitors in the Etiology of Myasthenia Gravis and Lambert-Eaton Myasthenic Syndrome: A Systematic Review. Front. Neurol. 2022, 13, 1004810. [Google Scholar] [CrossRef]
- Young, A.; Quandt, Z.; Bluestone, J.A. The Balancing Act between Cancer Immunity and Autoimmunity in Response to Immunotherapy. Cancer Immunol. Res. 2018, 6, 1445–1452. [Google Scholar] [CrossRef]
- Hoos, A.; Eggermont, A.M.M.; Janetzki, S.; Hodi, F.S.; Ibrahim, R.; Anderson, A.; Humphrey, R.; Blumenstein, B.; Old, L.; Wolchok, J. Improved Endpoints for Cancer Immunotherapy Trials. J. Natl. Cancer Inst. 2010, 102, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 93–110. ISBN 978-1-4939-3578-9. [Google Scholar]
- Rapier-Sharman, N.; Clancy, J.; Pickett, B.E. Joint Secondary Transcriptomic Analysis of Non-Hodgkin’s B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers. J. Bioinforma. Syst. Biol. 2022, 5, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Faramand, R.; Jain, M.; Staedtke, V.; Kotani, H.; Bai, R.; Reid, K.; Lee, S.B.; Spitler, K.; Wang, X.; Cao, B.; et al. Tumor Microenvironment Composition and Severe Cytokine Release Syndrome (CRS) Influence Toxicity in Patients with Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4823–4831. [Google Scholar] [CrossRef] [PubMed]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.-W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer Sequence Variants and Transcription-Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B-Cell Lymphoma. Immunity 2015, 42, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.; Kretzmer, L.Z.; Koues, O.I.; Payton, J.E.; Oltz, E.M.; Cashen, A.; Polic, B.; Schreiber, R.D.; Shaw, A.S.; Markiewicz, M.A. NKG2D-NKG2D Ligand Interaction Inhibits the Outgrowth of Naturally Arising Low-Grade B Cell Lymphoma In Vivo. J. Immunol. 2016, 196, 4805–4813. [Google Scholar] [CrossRef] [PubMed]
- Teater, M.; Dominguez, P.M.; Redmond, D.; Chen, Z.; Ennishi, D.; Scott, D.W.; Cimmino, L.; Ghione, P.; Chaudhuri, J.; Gascoyne, R.D.; et al. AICDA Drives Epigenetic Heterogeneity and Accelerates Germinal Center-Derived Lymphomagenesis. Nat. Commun. 2018, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-Cell Lymphomas in Patients with Myelofibrosis Receiving JAK1/2 Inhibitor Therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-Oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef]
- Rouhigharabaei, L.; Finalet Ferreiro, J.; Tousseyn, T.; van der Krogt, J.-A.; Put, N.; Haralambieva, E.; Graux, C.; Maes, B.; Vicente, C.; Vandenberghe, P.; et al. Non-IG Aberrations of FOXP1 in B-Cell Malignancies Lead to an Aberrant Expression of N-Truncated Isoforms of FOXP1. PLoS ONE 2014, 9, e85851. [Google Scholar] [CrossRef]
- Panwar, B.; Schmiedel, B.J.; Liang, S.; White, B.; Rodriguez, E.; Kalunian, K.; McKnight, A.J.; Soloff, R.; Seumois, G.; Vijayanand, P.; et al. Multi-Cell Type Gene Coexpression Network Analysis Reveals Coordinated Interferon Response and Cross-Cell Type Correlations in Systemic Lupus Erythematosus. Genome Res. 2021, 31, 659–676. [Google Scholar] [CrossRef]
- Andreoletti, G.; Lanata, C.M.; Trupin, L.; Paranjpe, I.; Jain, T.S.; Nititham, J.; Taylor, K.E.; Combes, A.J.; Maliskova, L.; Ye, C.J.; et al. Transcriptomic Analysis of Immune Cells in a Multi-Ethnic Cohort of Systemic Lupus Erythematosus Patients Identifies Ethnicity- and Disease-Specific Expression Signatures. Commun. Biol. 2021, 4, 488. [Google Scholar] [CrossRef]
- Andrews, J.M.; Pyfrom, S.C.; Schmidt, J.A.; Koues, O.I.; Kowalewski, R.A.; Grams, N.R.; Sun, J.J.; Berman, L.R.; Duncavage, E.J.; Lee, Y.-S.; et al. Loss of Synergistic Transcriptional Feedback Loops Drives Diverse B-Cell Cancers. EBioMedicine 2021, 71, 103559. [Google Scholar] [CrossRef]
- Shangguan, S.; Ehrenberg, P.K.; Geretz, A.; Yum, L.; Kundu, G.; May, K.; Fourati, S.; Nganou-Makamdop, K.; Williams, L.D.; Sawant, S.; et al. Monocyte-Derived Transcriptome Signature Indicates Antibody-Dependent Cellular Phagocytosis as a Potential Mechanism of Vaccine-Induced Protection against HIV-1. eLife 2021, 10, e69577. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Ren, Z.; Zhang, B.; Mao, L.; Zhu, G.; Gao, L.; Su, J.; Ye, J.; Long, Z.; Zhu, Y.; et al. Insufficient Epitope-Specific T Cell Clones Are Responsible for Impaired Cellular Immunity to Inactivated SARS-CoV-2 Vaccine in Older Adults. Nat. Aging 2023, 3, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, C.; Zhao, K.; Yang, Y.; Rassadkina, Y.; Fajnzylber, J.; Regan, J.; Li, J.Z.; Lichterfeld, M.; Yu, X.G. Immune-Profiling of SARS-CoV-2 Viremic Patients Reveals Dysregulated Innate Immune Responses. Front. Immunol. 2022, 13, 984553. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Wang, X.; Campbell, M.R.; Cho, H.-Y.; Pittman, G.S.; Martos, S.N.; Bell, D.A. Epigenomic Profiling of Isolated Blood Cell Types Reveals Highly Specific B Cell Smoking Signatures and Links to Disease Risk. Clin. Epigenet. 2023, 15, 90. [Google Scholar] [CrossRef]
- Rapier-Sharman, N.; Krapohl, J.; Beausoleil, E.J.; Gifford, K.T.L.; Hinatsu, B.R.; Hoffmann, C.S.; Komer, M.; Scott, T.M.; Pickett, B.E. Preprocessing of Public RNA-Sequencing Datasets to Facilitate Downstream Analyses of Human Diseases. Data 2021, 6, 75. [Google Scholar] [CrossRef]
- Clancy, J.; Hoffmann, C.S.; Pickett, B.E. Transcriptomics Secondary Analysis of Severe Human Infection with SARS-CoV-2 Identifies Gene Expression Changes and Predicts Three Transcriptional Biomarkers in Leukocytes. Comput. Struct. Biotechnol. J. 2023, 21, 1403–1413. [Google Scholar] [CrossRef]
- Gifford, K.T.L.; Pickett, B.E. Comparative Meta-Analysis of Host Transcriptional Response during Streptococcus Pneumoniae Carriage or Infection. Microb. Pathog. 2022, 173, 105816. [Google Scholar] [CrossRef]
- Moreno, C.; Bybee, E.; Tellez Freitas, C.M.; Pickett, B.E.; Weber, K.S. Meta-Analysis of Two Human RNA-Seq Datasets to Determine Periodontitis Diagnostic Biomarkers and Drug Target Candidates. Int. J. Mol. Sci. 2022, 23, 5580. [Google Scholar] [CrossRef]
- Ncbi/Sra-Tools 2024. Available online: https://github.com/ncbi/sra-tools (accessed on 9 September 2024).
- Orjuela, S.; Huang, R.; Hembach, K.M.; Robinson, M.D.; Soneson, C. ARMOR: An Automated Reproducible MOdular Workflow for Preprocessing and Differential Analysis of RNA-Seq Data. G3 Genes Genomes Genet. 2019, 9, 2089–2096. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake—A Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 7 June 2021).
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 June 2021).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Smyth, G.K. Camera: A Competitive Gene Set Test Accounting for Inter-Gene Correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Shaffer, J.P. Multiple Hypothesis Testing. Annu. Rev. Psychol. 1995, 46, 561–585. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-Scale Gene Function Analysis with the PANTHER Classification System. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Biotech. Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database J. Biol. Databases Curation 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.-H.; Chen, C.; Akyol, T.; Dusa, A.; Yu, G.; Cao, B.; Cai, P. ggVennDiagram: Intuitive Venn Diagram Software Extended. iMeta 2024, 3, e177. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting Systematic Drug-Target Identification and Prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef]
- Davis, J.; Handunnetti, S.M.; Sharpe, C.; Turner, G.; Anderson, M.A.; Roberts, A.W.; Seymour, J.F.; Tam, C.S.; Ritchie, D.; Koldej, R. Long Term Responses to Venetoclax and Ibrutinib in Mantle Cell Lymphoma Are Associated with Immunological Recovery and Prognostic Changes in Inflammatory Biomarkers. Blood 2019, 134, 2791. [Google Scholar] [CrossRef]
- Guo, J.; Gao, Y.; Wang, Y.; Zou, Y.; Du, Y.; Luo, C.; Shi, Y.; Yang, Y.; Wu, X.; Su, Y.; et al. Investigation of C1-Complex Regions Reveals New C1Q Variants Associated with Protection from Systemic Lupus Erythematosus, and Affect Its Transcript Abundance. Sci. Rep. 2018, 8, 8048. [Google Scholar] [CrossRef]
- Sontheimer, R.D.; Racila, E.; Racila, D.M. C1q: Its Functions within the Innate and Adaptive Immune Responses and Its Role in Lupus Autoimmunity. J. Investig. Dermatol. 2005, 125, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Suomela, S.; Cao, L.; Bowcock, A.; Saarialho-Kere, U. Interferon α-Inducible Protein 27 (IFI27) Is Upregulated in Psoriatic Skin and Certain Epithelial Cancers. J. Investig. Dermatol. 2004, 122, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Sagou, K.; Sato, Y.; Okuno, Y.; Watanabe, T.; Inagaki, T.; Motooka, Y.; Toyokuni, S.; Murata, T.; Kiyoi, H.; Kimura, H. Epstein-Barr Virus Lytic Gene BNRF1 Promotes B-Cell Lymphomagenesis via IFI27 Upregulation. PLoS Pathog. 2024, 20, e1011954. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole-Blood Transcriptional Profiling of Interferon-Inducible Genes Identifies Highly Upregulated IFI27 in Primary Myelofibrosis. Eur. J. Haematol. 2011, 87, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, L.; Wang, J.; Zhang, M.; Song, Z.; Ni, B.; You, Y. Identification of Key Biomarkers and Immune Infiltration in Systemic Lupus Erythematosus by Integrated Bioinformatics Analysis. J. Transl. Med. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.M.; Roderick, J.E.; Tan, S.H.; Tan, T.K.; Murphy, L.; Yu, J.; Li, R.; O’Connor, K.W.; Zhu, J.; Green, M.R.; et al. ESRRB Regulates Glucocorticoid Gene Expression in Mice and Patients with Acute Lymphoblastic Leukemia. Blood Adv. 2020, 4, 3154–3168. Available online: https://pubmed.ncbi.nlm.nih.gov/32658986/ (accessed on 7 June 2021). [PubMed]
- Dobbs Spendlove, M.; Gibson, T.M.; McCain, S.; Stone, B.C.; Gill, T.; Pickett, B.E. Pathway2Targets: An Open-Source Pathway-Based Approach to Repurpose Therapeutic Drugs and Prioritize Human Targets. PeerJ 2023, 11, e16088. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Liu, X.; Ji, J.; Forsti, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Autoimmune Disease and Subsequent Urological Cancer. J. Urol. 2013, 189, 2262–2268. [Google Scholar] [CrossRef]
- Hemminki, K.; Liu, X.; Ji, J.; Sundquist, J.; Sundquist, K. Autoimmune Disease and Subsequent Digestive Tract Cancer by Histology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 927–933. [Google Scholar] [CrossRef]
- Hemminki, K.; Liu, X.; Ji, J.; Sundquist, K.; Sundquist, J. Subsequent COPD and Lung Cancer in Patients with Autoimmune Disease. Eur. Respir. J. 2011, 37, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-D.; Das, R.; Goduni, L.; McClellan, S.; Hazlett, L.D.; Mahabeleshwar, G.H. Kruppel-like Factor 6 Promotes Macrophage-Mediated Inflammation by Suppressing B Cell Leukemia/Lymphoma 6 Expression. J. Biol. Chem. 2016, 291, 21271–21282. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the Autoimmune Disease Scleroderma with an Immunologic Response to Cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef]
- Shiokawa, M.; Kodama, Y.; Yoshimura, K.; Kawanami, C.; Mimura, J.; Yamashita, Y.; Asada, M.; Kikuyama, M.; Okabe, Y.; Inokuma, T.; et al. Risk of Cancer in Patients With Autoimmune Pancreatitis. Off. J. Am. Coll. Gastroenterol. ACG 2013, 108, 610–617. [Google Scholar] [CrossRef]
- Rahat, M.A.; Coffelt, S.B.; Granot, Z.; Muthana, M.; Amedei, A. Macrophages and Neutrophils: Regulation of the Inflammatory Microenvironment in Autoimmunity and Cancer. Mediat. Inflamm. 2016, 2016, e5894347. [Google Scholar] [CrossRef]
- Jiménez-Morales, S.; Ren, X.; Dean, M. Editorial: The Genetic Causes Underlying Immune Mediated Disease: A Focus on Autoimmunity and Cancer. Front. Genet. 2022, 13, 889160. [Google Scholar] [CrossRef]
- DePeaux, K.; Rivadeneira, D.B.; Lontos, K.; Dean, V.G.; Gunn, W.G.; Watson, M.J.; Yao, T.; Wilfahrt, D.; Hinck, C.; Wieteska, L.; et al. An Oncolytic Virus-Delivered TGFβ Inhibitor Overcomes the Immunosuppressive Tumor Microenvironment. J. Exp. Med. 2023, 220, e20230053. [Google Scholar] [CrossRef] [PubMed]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in Cancer Biology and Therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Demers-Mathieu, V. Optimal Selection of IFN-α-Inducible Genes to Determine Type I Interferon Signature Improves the Diagnosis of Systemic Lupus Erythematosus. Biomedicines 2023, 11, 864. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Phenotype | Type of Sequencing Reads | GEO Identifier | Number of Relevant Samples Included in Current Study |
|---|---|---|---|
| large B-cell lymphoma | paired end | GSE153437 [31] | 25 |
| follicular lymphoma | paired end | * GSE62241 [32,33] | 10 |
| diffuse large B-cell lymphoma | paired end | GSE95013 [34] | 29 |
| B-cell lymphoma | single end | * GSE110219 [35] | 1 |
| diffuse large B-cell lymphoma | paired end | GSE130751 [36] | 63 |
| diffuse large B-cell lymphoma | paired end | GSE50514 [37] | 7 |
| lupus B-cells | single end | * GSE149050 [38] | 18 |
| lupus B-cells | paired end | GSE164457 [39] | 120 |
| healthy B-cells | paired end | GSE145842 [40] | 6 |
| healthy B-cells | single end | * GSE149050 [38] | 14 |
| healthy B-cells | paired end | GSE181859 [41] | 20 |
| healthy B-cells | paired end | * GSE62241 [32,33] | 4 |
| healthy B-cells | single end | * GSE110219 [35] | 1 |
| healthy B-cells | paired end | GSE191088 [42] | 6 |
| healthy B-cells | paired end | GSE199868 (currently unpublished) | 13 |
| healthy B-cells | paired end | GSE216529 [43] | 2 |
| healthy B-cells | single end | GSE219888 [44] | 2 |
| healthy B-cells | paired end | GSE220113 [45] | 17 |
| healthy B-cells | single end | GSE222862 (currently unpublished) | 3 |
| Gene Symbol | Lupus * log2FC | Lupus * FDR | Lymphoma ** log2FC | Lymphoma ** FDR | IIT Score | IIT Corrected p-Value *** | |
|---|---|---|---|---|---|---|---|
| C+A+ Quad I | TPM2 | 1.85 | 3.25 × 10−8 | 3.88 | 1.65 × 10−25 | 21.81 | 0 |
| PTMS | 2.03 | 2.05 × 10−10 | 3.72 | 8.08 × 10−27 | 20.21 | 0 | |
| PLXNA1 | 1.11 | 3.14 × 10−12 | 2.75 | 1.04 × 10−34 | 19.35 | 0 | |
| SIX5 | 1.72 | 5.53 × 10−12 | 2.79 | 7.36 × 10−34 | 18.76 | 0 | |
| C+A− Quad II | LGALS3BP | −1.57 | 3.57 × 10−10 | 3.55 | 3.62 × 10−36 | 27.08 | 0 |
| VSIG4 | −1.65 | 5.81 × 10−8 | 6.13 | 4.83 × 10−29 | 17.62 | 0 | |
| FKBP5 | −1.33 | 1.77 × 10−9 | 2.25 | 8.87 × 10−36 | 16.57 | 0 | |
| ARMCX1 | −1.19 | 8.57 × 10−7 | 2.87 | 3.10 × 10−22 | 12.87 | 0 | |
| C−A− Quad III | MED30 | −0.895 | 1.54 × 10−12 | −1.46 | 5.07 × 10−41 | 19.64 | 0 |
| ING3 | −0.983 | 1.58 × 10−17 | −1.63 | 3.96 × 10−40 | 18.15 | 0 | |
| OSER1 | −0.752 | 6.09 × 10−14 | −1.6 | 8.45 × 10−50 | 17.44 | 0 | |
| PLD4 | −1.05 | 2.23 × 10−10 | −3.5 | 6.43 × 10−26 | 17.12 | 0 | |
| C−A+ Quad IV | PNRC1 | 1.21 | 5.07 × 10−16 | −2.11 | 1.16 × 10−43 | 28.32 | 0 |
| SLC12A6 | 1.04 | 6.91 × 10−15 | −2.46 | 3.48 × 10−37 | 21.8 | 0 | |
| OTUD1 | 1.47 | 7.92 × 10−18 | −2.24 | 5.46 × 10−37 | 21.37 | 0 | |
| MARCH8 | 0.932 | 6.91 × 10−15 | −1.86 | 5.35 × 10−40 | 19.8 | 0 |
| Term | Overlap | Bonferroni p-Value | Odds Ratio | Combined Score | GO DAG * |
|---|---|---|---|---|---|
| RNA Binding (GO:0003723) | 542/1411 | 7.08 × 10−26 | 1.92 | 124.82 | Molecular Function |
| Cytoplasmic Translation (GO:0002181) | 42/93 | 3.05 × 10−16 | 8.33 | 366.05 | Biological Process |
| RNA Binding (GO:0003723) | 193/1411 | 1.36 × 10−11 | 1.95 | 61.83 | Molecular Function |
| Proton Motive Force-Driven ATP Synthesis (GO:0015986) | 26/60 | 4.07 × 10−10 | 8.99 | 268.95 | Biological Process |
| RNA Binding (GO:0003723) | 209/1411 | 5.27 × 10−10 | 1.82 | 50.73 | Molecular Function |
| Pathway | Overlap | FDR p-Value | Odds Ratio | Combined Score | Database |
|---|---|---|---|---|---|
| Neutrophil Degranulation | 197/468 | 1.54 × 10−15 | 2.17 | 74.13 | Reactome |
| Innate Immune System | 372/1035 | 1.06 × 10−14 | 1.69 | 54.48 | Reactome |
| Immune System | 636/1943 | 2.44 × 10−14 | 1.49 | 46.55 | Reactome |
| Metabolism Of RNA | 253/666 | 2.96 × 10−13 | 1.83 | 52.94 | Reactome |
| Cellular Responses To Stress | 268/722 | 1.22 × 10−12 | 1.77 | 48.52 | Reactome |
| Cellular Responses To Stimuli | 272/736 | 1.46 × 10−12 | 1.76 | 47.88 | Reactome |
| Metabolism Of Proteins | 608/1890 | 3.59 × 10−12 | 1.44 | 37.98 | Reactome |
| Transcriptional Regulation By TP53 * | 145/354 | 9.17 × 10−11 | 2.06 | 47.65 | Reactome |
| Translation | 120/281 | 1.77 × 10−10 | 2.21 | 49.64 | Reactome |
| Adaptive Immune System | 261/733 | 3.45 × 10−10 | 1.65 | 36.00 | Reactome |
| Gene Symbol | Quadrant | Immune Imbalance Score | IIT Corrected p-Value * | Number Unique Drugs | Number Approved Drugs | Weighted Target Score |
|---|---|---|---|---|---|---|
| MAP3K1 | Quad IV | 16.47 | 0 | 1 | 0 | 698 |
| NR1H2 | Quad III | 15.24 | 0 | 5 | 0 | 186 |
| KCNQ1 | Quad IV | 12.96 | 0 | 8 | 1 | 662 |
| GABBR1 | Quad IV | 11.8 | 0 | 7 | 1 | 320 |
| PIKFYVE | Quad IV | 11.79 | 0 | 2 | 0 | 123 |
| SIRT1 | Quad IV | 10.86 | 0 | 1 | 0 | 1219 |
| PTGER4 | Quad III | 9.64 | 0 | 4 | 1 | 438 |
| CCN2 | Quad I | 9.25 | 0 | 1 | 0 | 927.5 |
| CXCL10 | Quad II | 8.81 | 0 | 2 | 0 | 1212 |
| CD3G | Quad II | 8.29 | 0 | 11 | 1 | 195 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapier-Sharman, N.; Kim, S.; Mudrow, M.; Told, M.T.; Fischer, L.; Fawson, L.; Parry, J.; Poole, B.D.; O’Neill, K.L.; Piccolo, S.R.; et al. Comparison of B-Cell Lupus and Lymphoma Using a Novel Immune Imbalance Transcriptomics Algorithm Reveals Potential Therapeutic Targets. Genes 2024, 15, 1215. https://doi.org/10.3390/genes15091215
Rapier-Sharman N, Kim S, Mudrow M, Told MT, Fischer L, Fawson L, Parry J, Poole BD, O’Neill KL, Piccolo SR, et al. Comparison of B-Cell Lupus and Lymphoma Using a Novel Immune Imbalance Transcriptomics Algorithm Reveals Potential Therapeutic Targets. Genes. 2024; 15(9):1215. https://doi.org/10.3390/genes15091215
Chicago/Turabian StyleRapier-Sharman, Naomi, Sehi Kim, Madelyn Mudrow, Michael T. Told, Lane Fischer, Liesl Fawson, Joseph Parry, Brian D. Poole, Kim L. O’Neill, Stephen R. Piccolo, and et al. 2024. "Comparison of B-Cell Lupus and Lymphoma Using a Novel Immune Imbalance Transcriptomics Algorithm Reveals Potential Therapeutic Targets" Genes 15, no. 9: 1215. https://doi.org/10.3390/genes15091215
APA StyleRapier-Sharman, N., Kim, S., Mudrow, M., Told, M. T., Fischer, L., Fawson, L., Parry, J., Poole, B. D., O’Neill, K. L., Piccolo, S. R., & Pickett, B. E. (2024). Comparison of B-Cell Lupus and Lymphoma Using a Novel Immune Imbalance Transcriptomics Algorithm Reveals Potential Therapeutic Targets. Genes, 15(9), 1215. https://doi.org/10.3390/genes15091215