
Transcriptomic Evidence Reveals the Dysfunctional Mechanism of Synaptic Plasticity Control in ASD


Highlights
- We established a workflow to convert signal transduction networks into mRNA regulatory networks.
- We developed a Boolean regulatory network model tailored to single-cell data analysis.
- We designed a probabilistic model for single-cell data interpretation.
- Our approach contributes to the investigation of convergent causal molecular mechanisms in autism.
- Our novel networks and models can be broadly applied to other diseases and computational biology research.
Abstract

1. Introduction
2. Materials and Methods
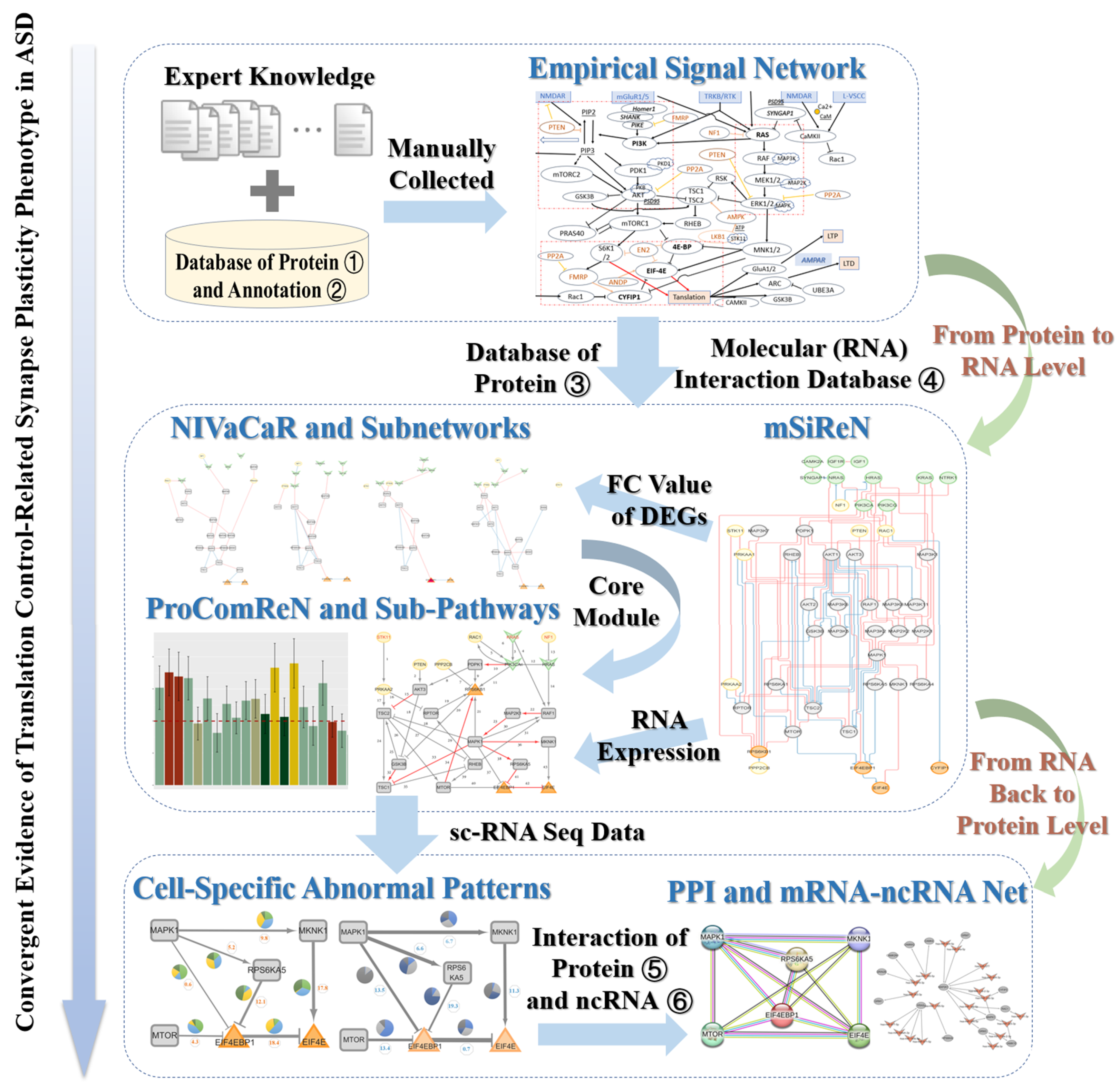
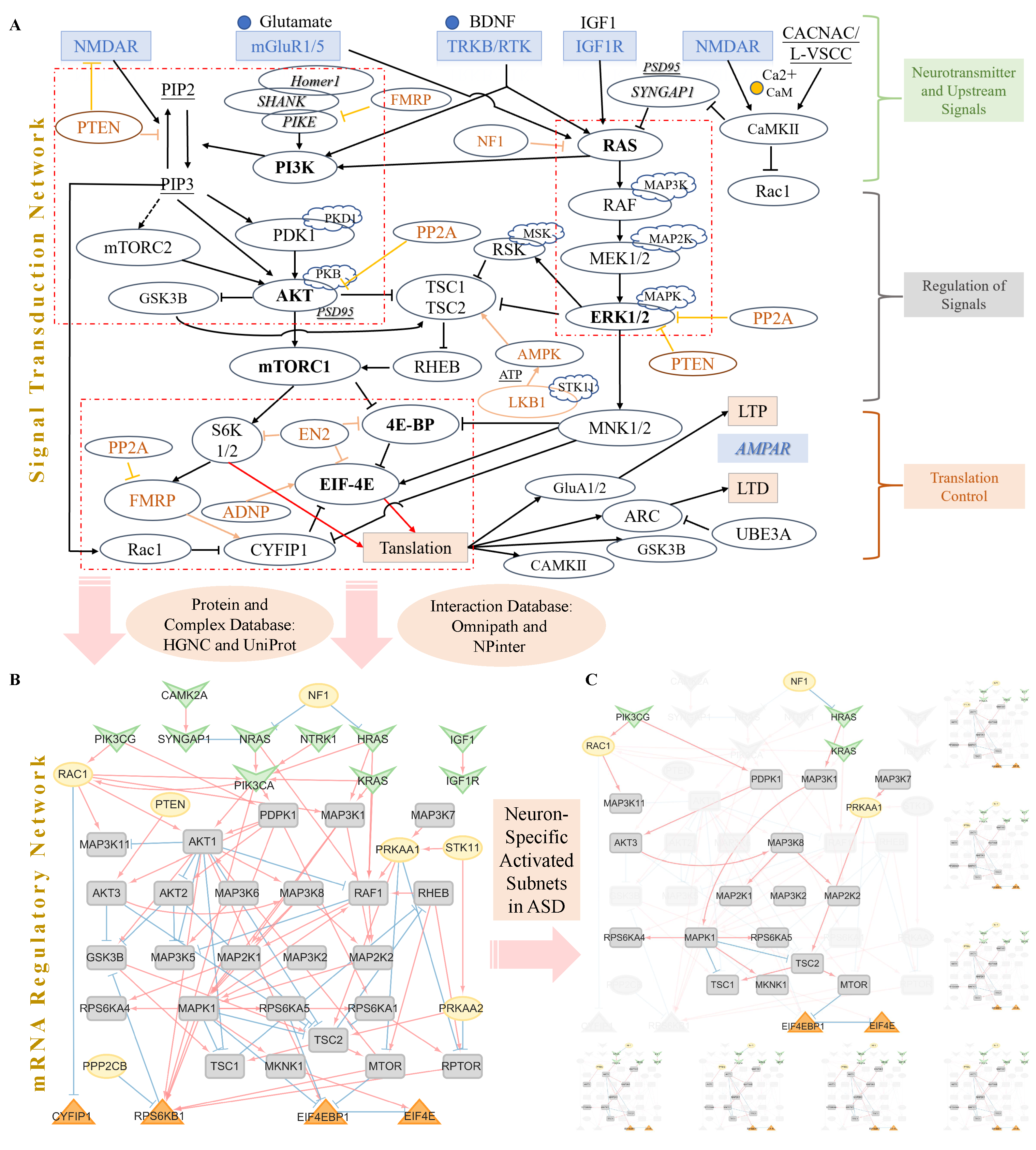
2.1. Signal Transduction Network About Translation Control of Postsynapse Plasticity
2.2. The Pipeline of Constructing the mSiReN
2.2.1. Initial Nodes and Final Nodes from Empirical Knowledge
2.2.2. Match with Annotation Database
2.2.3. Match with Interaction Database
- Selection of Signal Annotation Databases and Key Signaling Pathways: Initially, we match the nodes from the literature-mined network with annotation databases for signal pathway-related information.
- Matching the initial input and terminal nodes with other network nodes in the annotation databases to find molecular interaction relationships. Only nodes matched with signal annotation databases from the literature-mined network are included in the final network. Matching involves identifying upstream signaling and neurotransmitter receptor nodes in the source nodes of the database, as well as translation control nodes in the target nodes. Other signal nodes are also matched.
- Choosing the direct interactions of signal nodes: Searching for regulatory subnetworks in the signal-regulated network containing the nodes of interest.
2.3. The High-Throughput scRNA-Seq Data and the Preprocessing of DEGs
2.3.1. Single-Cell RNA Sequencing Data
2.3.2. Differential Expression Analysis
2.4. CS-NIVaCaR: Cell-Specific Network Inference via Integer Value Programming and Causal Reasoning
The Objective Function for NIVaCaR
2.5. CS-ProComReN: Cell-Specific Probabilistic Contextualization for mRNA Signaling Regulatory Networks
- The Procedure of the ProComReN Algorithm
- The Steps of the ProComReN Algorithm
- I.
- Model Initialization:
- Providing the precursor RNA network as PKN.
- The combination of the activity/inhibitor of input nodes as the experimental conditions.
- The RNA expression of molecules as node measurements.
- Initialize the normally distributed values of the nodes, except for the input nodes.
- Assign random initial weights to the edges.
- II.
- Computation of Steady-State:
- Update the values of the nodes iteratively according to the DBN formulation:
- Compute the expected value of each node’s probability distribution based on the values of its parent nodes and the associated weights.
- III.
- Contextualization with Experimental Data:
- Compare the Mean Squared Error (MSE) between the estimated and normalized measured values.
- Define an objective function:
- Use a gradient-descent algorithm (e.g., fmincon, function minimization with constraints, with the interior-point method) to optimize by adjusting the weights.
- Iterate the optimization process until convergence or a stopping criterion is met.
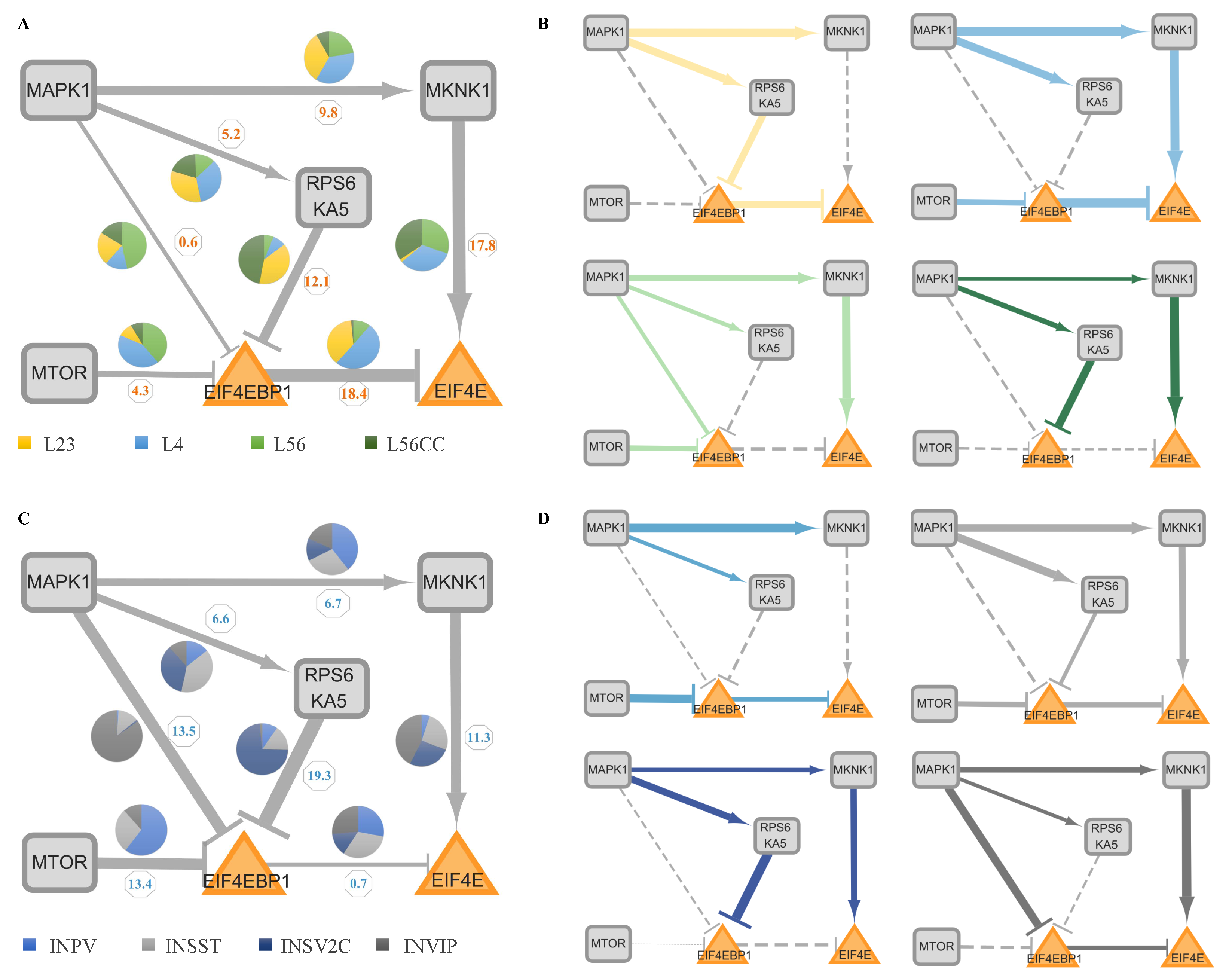
2.6. The Strength of the Sub-Pathway (SSP) and Abnormality Index of the Sub-Pathway (AISP)
2.7. The Framework of Analysis
2.8. Computational Packages and Database
3. Results
3.1. Signal Transduction Network and mSiReN
3.1.1. Signal Network of Glutamate Synaptic Plasticity
3.1.2. The Construction Pipeline of mSiReN
3.2. The Cell-Type-Specific Activated Sub-Networks and NIVaCaR
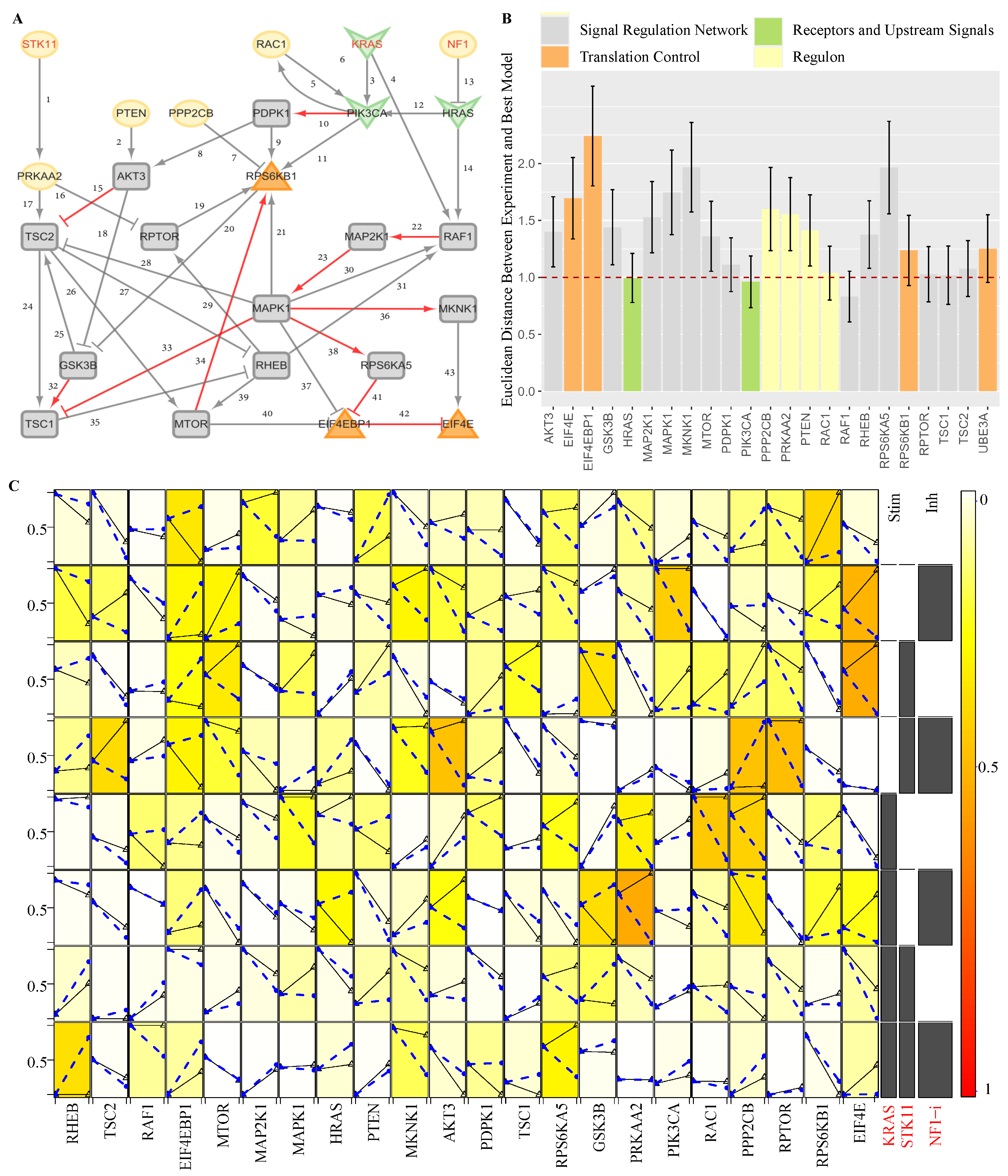
3.3. The Activated Sub-Pathways and ProComReN
3.3.1. The ProComReN Results
3.3.2. The Activated Sub-Pathways
3.3.3. The Evaluation of Activated Sub-Pathways
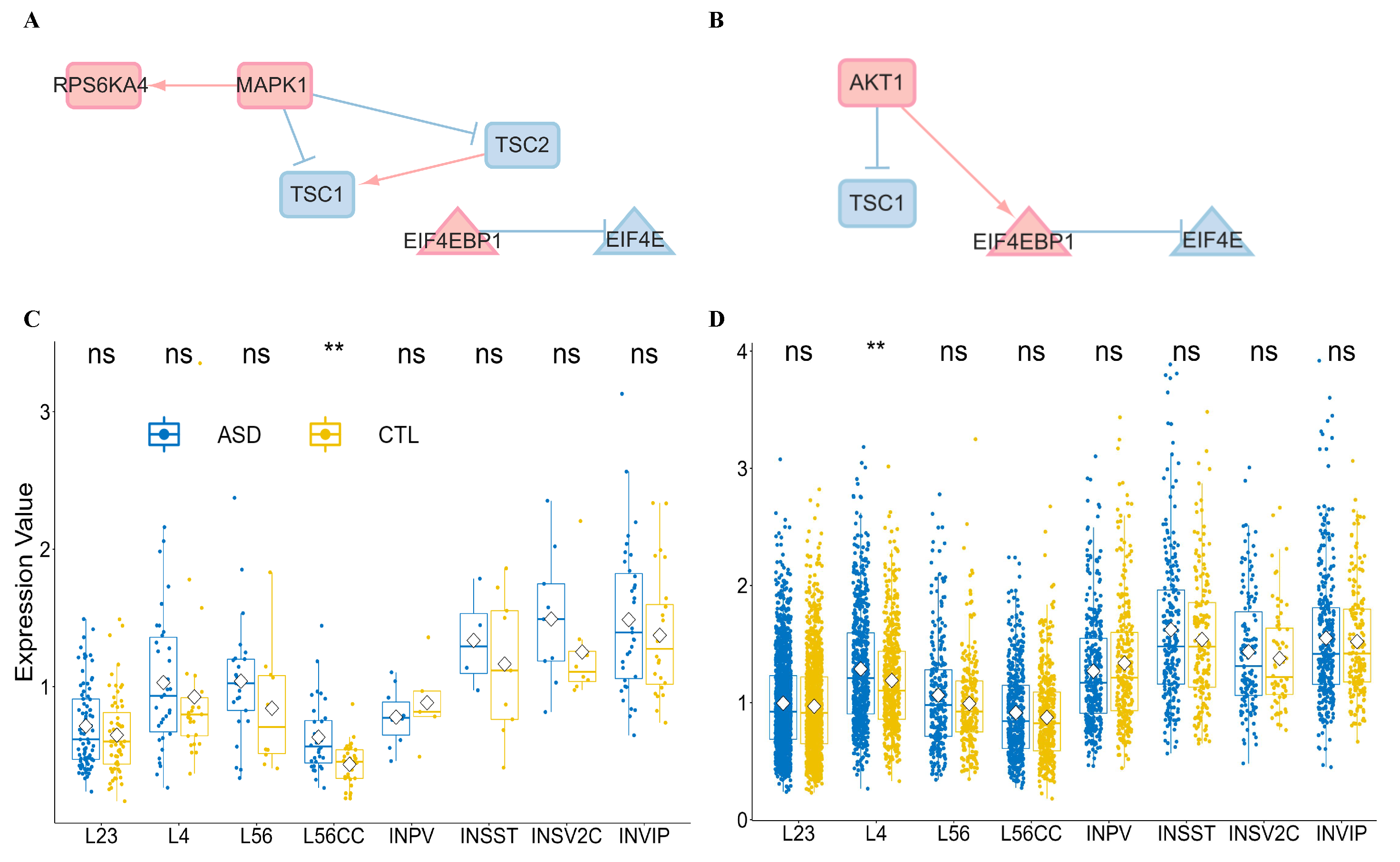
3.4. Convergent Evidence on Translation Control of Synaptic Plasticity
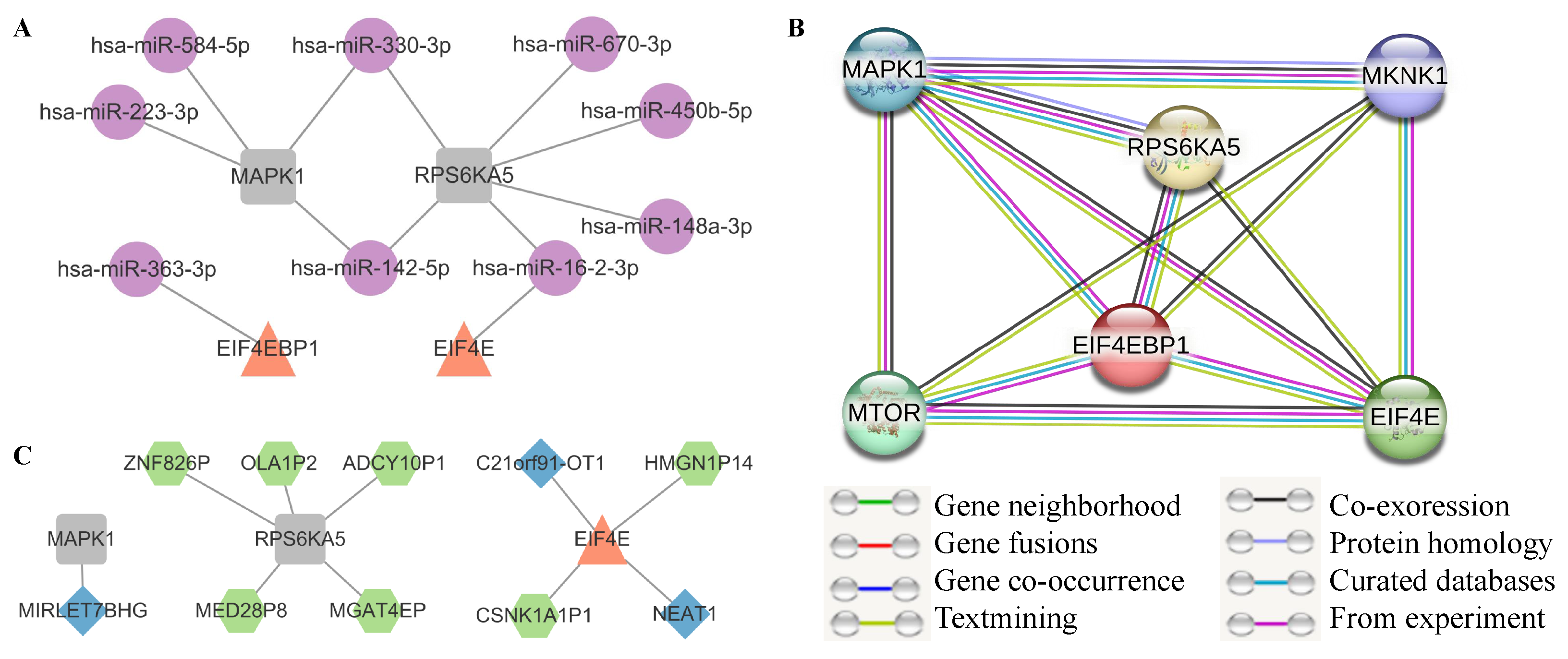
3.4.1. Convergence in Abnormal Non-Coding RNAs and Pseudogenes
3.4.2. The Reliability of Edges with Protein Interaction in mSiReN
3.4.3. Core Network with ADRI Score
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lieberman, J.A.; First, M.B. Psychotic disorders. N. Engl. J. Med. 2018, 379, 270–280. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30021088 (accessed on 13 June 2023). [CrossRef]
- Sullivan, P.F.; Geschwind, D.H. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell 2019, 177, 162–183. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30901538 (accessed on 12 June 2023). [CrossRef] [PubMed]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental disorders: From genetics to functional pathways. Trends Neurosci. 2020, 43, 608–621. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32507511 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Courchesne, E.; Gazestani, V.H.; Lewis, N.E. Prenatal origins of asd: The when, what, and how of asd development. Trends Neurosci. 2020, 43, 326–342. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32353336 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Lord, C.; S. Brugha, T.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.; Jones, R.; Pickles, A.; Matthew, S.; et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 2020, 6, 5. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31949163 (accessed on 13 June 2023). [CrossRef]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; et al. Environmental risk factors and biomarkers for autism spectrum disorder: An umbrella review of the evidence. Lancet Psychiatry 2019, 6, 590–600. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31230684 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25891009 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the cores of autism. Cell 2019, 178, 1287–1298. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31491383 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Geschwind, D.H. Advances in autism. Annu. Rev. Med. 2009, 60, 367–380. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19630577 (accessed on 13 June 2023). [CrossRef]
- Hirota, T.; King, B.H. Autism spectrum disorder: A review. JAMA 2023, 329, 157–168. [Google Scholar] [CrossRef]
- Buch, A.M.; Vértes, P.E.; Seidlitz, J.; Kim, S.H.; Grosenick, L.; Liston, C. Molecular and network-level mechanisms explaining individual differences in autism spectrum disorder. Nat. Neurosci. 2023, 26, 650–663. [Google Scholar] [CrossRef]
- Roehr, B. American psychiatric association explains dsm-5. BMJ 2013, 346, f3591. [Google Scholar] [CrossRef]
- Christensen, D.L. Prevalence and characteristics of autism spectrum disorder among children aged 4 years—Early autism and developmental disabilities monitoring network, seven sites, united states, 2010, 2012, and 2014. MMWR Surveill. Summ. 2019, 68, 1–19. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30973853 (accessed on 13 June 2023). [CrossRef]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism. Dev. Disord. 1994, 24, 659–685. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7814313 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Jeste, S.S.; Geschwind, D.H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 2014, 10, 74–81. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24468882 (accessed on 13 June 2023). [CrossRef]
- Chen, J.A.; Penagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. 2015, 10, 111–144. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25621659 (accessed on 13 June 2023). [CrossRef] [PubMed]
- de la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27050589 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18258309 (accessed on 13 June 2023). [CrossRef]
- Stoner, R. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24670167 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19896929 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Courchesne, E. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J., 3rd; Bear, M.F. The autistic neuron: Troubled translation? Cell 2008, 135, 401–406. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18984149 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Isshiki, M. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun. 2014, 5, 4742. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26289574 (accessed on 13 June 2023). [CrossRef]
- Bonsi, P.; De Jaco, A.; Fasano, L.; Gubellini, P. Postsynaptic autism spectrum disorder genes and synaptic dysfunction. Neurobiol. Dis. 2022, 162, 105564. Available online: https://www.ncbi.nlm.nih.gov/pubmed/34838666 (accessed on 13 June 2023). [CrossRef]
- Andrianova, L.; Yanakieva, S.; Margetts-Smith, G.; Kohli, S.; Brady, E.S.; Aggleton, J.P.; Craig, M.T. No evidence from complementary data sources of a direct glutamatergic projection from the mouse anterior cingulate area to the hippocampal formation. eLife 2023, 12, e77364. [Google Scholar] [CrossRef] [PubMed]
- Yap, E.L.; Greenberg, M.E. Activity-regulated transcription: Bridging the gap between neural activity and behavior. Neuron 2018, 100, 330–348. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30359600 (accessed on 13 June 2023). [CrossRef]
- Edfawy, M.; Guedes, J.R.; Pereira, M.I.; Laranjo, M.; Carvalho, M.J.; Ferreira, P.A. Abnormal mglur-mediated synaptic plasticity and autism-like behaviours in gprasp2 mutant mice. Nat. Commun. 2019, 10, 1431. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Zukin, R.S. A synaptic perspective of fragile x syndrome and autism spectrum disorders. Neuron 2019, 101, 1070–1088. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30897358 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Sacai, H.; Sakoori, K.; Konno, K.; Suzuki, H.; Watanabe, T.; Uesaka, N.; Kano, M. Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun. 2020, 11, 5140. [Google Scholar] [CrossRef]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23325215 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 2019, 101, 648–661.e4. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30679017 (accessed on 13 June 2023). [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015, 87, 684–698. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26291155 (accessed on 13 June 2023). [CrossRef]
- Lee, E.; Lee, J.; Kim, E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 2017, 81, 838–847. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27450033 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Chamberland, S. Brief synaptic inhibition persistently interrupts firing of fast-spiking interneurons. Neuron 2023, 111, 1264–1281.e5. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-generation dna sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; Buxbaum, J.D. Genetics and genomics of autism spectrum disorder: Embracing complexity. Hum. Mol. Genet. 2015, 24, R24–R31. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26188008 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Zito, A.; Reuveni, E.; Elliott, E. Dysregulation of cortical neuron dna methylation profile in autism spectrum disorder. Cereb. Cortex. 2017, 27, 5739–5754. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29028941 (accessed on 13 June 2023). [CrossRef]
- Satterstrom, F.K. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 2019, 22, 1961–1965. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31768057 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Satterstrom, F.K. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584.e23. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31981491 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Trost, B. Genome-wide detection of tandem dna repeats that are expanded in autism. Nature 2020, 586, 80–86. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32717741 (accessed on 13 June 2023). [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Lein, E.S.; Belgard, T.G.; Hawrylycz, M.; Molnar, Z. Transcriptomic perspectives on neocortical structure, development, evolution, and disease. Annu. Rev. Neurosci. 2017, 40, 629–652. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28661727 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Hernandez, L.M. Transcriptomic insight into the polygenic mechanisms underlying psychiatric disorders. Biol. Psychiatry 2021, 89, 54–64. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32792264 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Gandal, M.J. Broad transcriptomic dysregulation occurs across the cerebral cortex in asd. Nature 2022, 611, 532–539. Available online: https://www.ncbi.nlm.nih.gov/pubmed/36323788 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Ament, S.A.; Poulopoulos, A. The brain’s dark transcriptome: Sequencing rna in distal compartments of neurons and glia. Curr. Opin. Neurobiol. 2023, 81, 102725. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.C. Buffering of transcription rate by mrna half-life is a conserved feature of rett syndrome models. Nat. Commun. 2023, 14, 1896. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18414403 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Krumm, N. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25961944 (accessed on 13 June 2023). [CrossRef]
- Luo, W.; Zhang, C.; Jiang, Y.H.; Brouwer, C.R. Systematic reconstruction of autism biology from massive genetic mutation profiles. Sci. Adv. 2018, 4, e1701799. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29651456 (accessed on 13 June 2023). [CrossRef] [PubMed]
- RK, C.Y. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28263302 (accessed on 13 June 2023).
- Havdahl, A. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. Available online: https://www.ncbi.nlm.nih.gov/pubmed/33634770 (accessed on 13 June 2023). [CrossRef]
- LaSalle, J.M. Epigenomic signatures reveal mechanistic clues and predictive markers for autism spectrum disorder. Mol. Psychiatry 2023, 28, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.E.; Parikshak, N.N.; Belgard, T.G.; Geschwind, D.H. Genome-wide, integrative analysis implicates microrna dysregulation in autism spectrum disorder. Nat. Neurosci. 2016, 19, 1463–1476. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27571009 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Velmeshev, D. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Iossifov, I. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25363768 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Sanders, S.J. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26402605 (accessed on 13 June 2023). [CrossRef]
- Willsey, H.R.; Willsey, A.J.; Wang, B.; State, M.W. Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat. Rev. Neurosci. 2022, 23, 323–341. Available online: https://www.ncbi.nlm.nih.gov/pubmed/35440779 (accessed on 13 June 2023). [CrossRef]
- Quesnel-Vallieres, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism spectrum disorder: Insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30390048 (accessed on 13 June 2023). [CrossRef]
- Willsey, A.J. The psychiatric cell map initiative: A convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 2018, 174, 505–520. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30053424 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Sestan, N.; State, M.W. Lost in translation: Traversing the complex path from genomics to therapeutics in autism spectrum disorder. Neuron 2018, 100, 406–423. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30359605 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Searles Quick, V.B.; Wang, B.; State, M.W. Leveraging large genomic datasets to illuminate the pathobiology of autism spectrum disorders. Neuropsychopharmacology 2021, 46, 55–69. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32668441 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Voineagu, I. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21614001 (accessed on 13 June 2023). [CrossRef]
- Mahony, C.; O’Ryan, C. Convergent canonical pathways in autism spectrum disorder from proteomic, transcriptomic and dna methylation data. Int. J. Mol. Sci. 2021, 22, 10757. Available online: https://www.ncbi.nlm.nih.gov/pubmed/34639097 (accessed on 13 June 2023). [CrossRef]
- Ramaswami, G. Integrative genomics identifies a convergent molecular subtype that links epigenomic with transcriptomic differences in autism. Nat. Commun. 2020, 11, 4873. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32978376 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Noel, J.-P.; Angelaki, D.E. A theory of autism bridging across levels of description. Trends Cogn. Sci. 2023, 27, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Iannuccelli, M. Curation of causal interactions mediated by genes associated with autism accelerates the understanding of gene-phenotype relationships underlying neurodevelopmental disorders. Mol. Psychiatry 2023, 29, 186–196. Available online: https://www.ncbi.nlm.nih.gov/pubmed/38102483 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Melas, I.N. Identification of drug-specific pathways based on gene expression data: Application to drug induced lung injury. Integr. Biol. 2015, 7, 904–920. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25932872 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Liu, A. From expression footprints to causal pathways: Contextualizing large signaling networks with carnival. NPJ Syst. Biol. Appl. 2019, 5, 40. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31728204 (accessed on 13 June 2023). [CrossRef]
- Szalai, B.; Saez-Rodriguez, J. Why do pathway methods work better than they should? FEBS Lett. 2020, 594, 4189–4200. Available online: https://www.ncbi.nlm.nih.gov/pubmed/33270910 (accessed on 13 June 2023). [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29953871 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Nussinov, R. Neurodevelopmental disorders, like cancer, are connected to impaired chromatin remodelers, pi3k/mtor, and pak1-regulated mapk. Biophys. Rev. 2023, 15, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Moya-Alvarado, G. Bdnf/trkb signaling endosomes in axons coordinate creb/mtor activation and protein synthesis in the cell body to induce dendritic growth in cortical neurons. eLife 2023, 12, e77455. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I. The fragile x syndrome protein represses activity-dependent translation through cyfip1, a new 4e-bp. Cell 2008, 134, 1042–1054. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18805096 (accessed on 13 June 2023). [CrossRef]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A cerna hypothesis: The rosetta stone of a hidden rna language? Cell 2011, 146, 353–358. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21802130 (accessed on 13 June 2023). [CrossRef]
- Zampa, F.; Hartzell, A.L.; Zolboot, N.; Lippi, G. Non-coding rnas: The gatekeepers of neural network activity. Curr. Opin. Neurobiol. 2019, 57, 54–61. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30743177 (accessed on 13 June 2023). [CrossRef]
- Geisler, S.; Coller, J. Rna in unexpected places: Long non-coding rna functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24105322 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding rnas. Nat. Cell Biol. 2019, 21, 542–551. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31048766 (accessed on 13 June 2023). [CrossRef]
- Liau, W.S.; Samaddar, S.; Banerjee, S.; Bredy, T.W. On the functional relevance of spatiotemporally-specific patterns of experience-dependent long noncoding rna expression in the brain. RNA Biol. 2021, 18, 1025–1036. Available online: https://www.ncbi.nlm.nih.gov/pubmed/33397182 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Cheetham, S.W.; Faulkner, G.J.; Dinger, M.E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 2020, 21, 191–201. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31848477 (accessed on 13 June 2023). [CrossRef]
- Tang, G. Loss of mtor-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25155956 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Pagani, M. mtor-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat. Commun. 2021, 12, 6084. [Google Scholar] [CrossRef]
- Gkogkas, C.G. Autism-related deficits via dysregulated eif4e-dependent translational control. Nature 2013, 493, 371–377. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23172145 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Panja, D. Two-stage translational control of dentate gyrus ltp consolidation is mediated by sustained bdnf-trkb signaling to mnk. Cell Rep. 2014, 9, 1430–1445. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25453757 (accessed on 13 June 2023). [CrossRef]
- Aguilar-Valles, A. Inhibition of group i metabotropic glutamate receptors reverses autistic-like phenotypes caused by deficiency of the translation repressor eif4e binding protein 2. J. Neurosci. 2015, 35, 11125–11132. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26245973 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Wiebe, S. The eif4e homolog 4ehp (eif4e2) regulates hippocampal long-term depression and impacts social behavior. Mol. Autism. 2020, 11, 92. Available online: https://www.ncbi.nlm.nih.gov/pubmed/33225984 (accessed on 13 June 2023). [CrossRef]
- Ruzzo, E.K. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178, 850–866.e26. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31398340 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Post, K.L.; Belmadani, M. Multi-model functionalization of disease-associated pten missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat. Commun. 2020, 11, 2073. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22411467 (accessed on 13 June 2023). [CrossRef] [PubMed]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using gene expression noise to understand gene regulation. Science 2012, 336, 183–187. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22499939 (accessed on 13 June 2023). [CrossRef]
- Jung, J.; Ohk, J.; Kim, H.; Holt, C.E.; Park, H.J.; Jung, H. mRNA transport, translation, and decay in adult mammalian central nervous system axons. Neuron 2023, 111, 650–668.e4. [Google Scholar] [CrossRef] [PubMed]
- Terfve, C.; Cokelaer, T.; Henriques, D.; MacNamara, A.; Goncalves, E.; Morris, M.K.; van Iersel, M.; Lauffenburger, D.A.; Saez-Rodriguez, J. Cellnoptr: A flexible toolkit to train protein signaling networks to data using multiple logic formalisms. BMC Syst. Biol. 2012, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Le Novere, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25645874 (accessed on 13 June 2023). [CrossRef]
- Morris, M.K.; Saez-Rodriguez, J.; Clarke, D.C.; Sorger, P.K.; Lauffenburger, D.A. Training signaling pathway maps to biochemical data with constrained fuzzy logic: Quantitative analysis of liver cell responses to inflammatory stimuli. PLoS Comput. Biol. 2011, 7, e1001099. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21408212 (accessed on 13 June 2023). [CrossRef]
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; Di Bernardo, D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007, 3, 78. [Google Scholar] [CrossRef]
- Li, F.; Long, T.; Lu, Y.; Ouyang, Q.; Tang, C. The yeast cell-cycle network is robustly designed. Proc. Natl. Acad. Sci. USA 2004, 101, 4781–4786. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.; Thieffry, D.; Alvarez-Buylla, E.R. Genetic control of flower morphogenesis in arabidopsis thaliana: A logical analysis. Bioinformatics 1999, 15, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Saez-Rodriguez, J.; Alexopoulos, L.G.; Epperlein, J.; Samaga, R.; Lauffenburger, D.A.; Klamt, S.; Sorger, P.K. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol. Syst. Biol. 2009, 5, 331. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S.A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 1969, 22, 437–467. [Google Scholar] [CrossRef]
- Trairatphisan, P.; Mizera, A.; Pang, J.; Tantar, A.A.; Schneider, J.; Sauter, T. Recent development and biomedical applications of probabilistic boolean networks. Cell Commun. Signal. 2013, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Lähdesmäki, H.; Hautaniemi, S.; Shmulevich, I.; Yli-Harja, O. Relationships between probabilistic boolean networks and dynamic bayesian networks as models of gene regulatory networks. Signal Process. 2006, 86, 814–834. [Google Scholar] [CrossRef] [PubMed]
- De Landtsheer, S.; Trairatphisan, P.; Lucarelli, P.; Sauter, T. Falcon: A toolbox for the fast contextualization of logical networks. Bioinformatics 2017, 33, 3431–3436. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28673016 (accessed on 13 June 2023). [CrossRef]
- Gjerga, E.; Trairatphisan, P.; Gabor, A.; Koch, H.; Chevalier, C.; Ceccarelli, F.; Dugourd, A.; Mitsos, A.; Saez-Rodriguez, J. Converting networks to predictive logic models from perturbation signalling data with cellnopt. Bioinformatics 2020, 36, 4523–4524. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32516357 (accessed on 13 June 2023). [CrossRef]
- Waltz, R.A.; Morales, J.L.; Nocedal, J.; Orban, D. An interior algorithm for nonlinear optimization that combines line search and trust region steps. Math. Program. 2006, 107, 391–408. [Google Scholar] [CrossRef]
- Coleman, T.F.; Li, Y. An interior trust region approach for nonlinear minimization subject to bounds. SIAM J. Optim. 1996, 6, 418–445. [Google Scholar] [CrossRef]
- Coleman, T.F.; Li, Y. On the convergence of interior-reflective newton methods for nonlinear minimization subject to bounds. Math. Program. 1994, 67, 189–224. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I: MAPK1 (->) MKNK1 (->) EIF4E | II: MAPK1 (->) RPS6KA5 (-|) EIF4EBP1 (-|) EIF4E | III: MAPK1 (-|) EIF4EBP1 (-|) EIF4E | IV: MTOR (-|) EIF4EBP1 (-|) EIF4E | Sub-Pathway Pattern (ASD) | |||||
|---|---|---|---|---|---|---|---|---|---|
| SSP | AISP | SSP | AISP | SSP | AISP | SSP | AISP | ||
| L23 | 4.1 | 31.4 | 32.7 | 33.69 | 9.8 | 12.7 | 7.7 | 11.89 | II |
| L4 | 82.81 | 34.72 | 10.11 | 29.72 | 8.55 | 10.97 | 45.6 | 21.14 | I and IV |
| L56 | 45.65 | 24.76 | 0.58 | 15.96 | 5.2 | 15.53 | 8.8 | 18.07 | I |
| L56CC | 19.74 | 25.25 | 1.01 | 23.45 | 0.3 | 15.51 | 0.27 | 15.39 | I |
| INPV | 9.9 | 21.47 | 1.12 | 13.02 | 0.33 | 9.29 | 27.72 | 20.55 | IV |
| INSST | 36.4 | 16.7 | 5.52 | 15.38 | 4.44 | 3.07 | 14.43 | 8.58 | I, II and IV |
| INSV2C | 18.56 | 11.71 | 10.66 | 15.34 | 0.17 | 3.92 | 0 | 3.76 | I |
| INVIP | 39.06 | 15.36 | 0.13 | 10.73 | 24.18 | 16.55 | 4.96 | 10.16 | I and III |
| Node1 | Node2 | Homology | Coexpr | Experimentally Determined Interaction | Database Annotated | Automated Textmining | Combined Score |
|---|---|---|---|---|---|---|---|
| EIF4E | MAPK1 | 0 | 0.062 | 0.127 | 0 | 0.438 | 0.5 |
| EIF4E | EIF4EBP1 | 0 | 0 | 0.996 | 0.9 | 0.994 | 0.999 |
| EIF4E | MTOR | 0 | 0.062 | 0.369 | 0.9 | 0.993 | 0.999 |
| EIF4E | MKNK1 | 0 | 0 | 0.637 | 0.9 | 0.833 | 0.993 |
| EIF4E | RPS6KA5 | 0 | 0.063 | 0 | 0 | 0.406 | 0.419 |
| EIF4EBP1 | MTOR | 0 | 0 | 0.982 | 0.9 | 0.913 | 0.999 |
| EIF4EBP1 | RPS6KA5 | 0 | 0.049 | 0.213 | 0.9 | 0.578 | 0.964 |
| EIF4EBP1 | MAPK1 | 0 | 0 | 0.485 | 0.8 | 0.438 | 0.937 |
| EIF4EBP1 | MKNK1 | 0 | 0.055 | 0 | 0 | 0.556 | 0.563 |
| MAPK1 | MKNK1 | 0.582 | 0.056 | 0.721 | 0.9 | 0.787 | 0.98 |
| MAPK1 | RPS6KA5 | 0.642 | 0.062 | 0.319 | 0.9 | 0.388 | 0.939 |
| MAPK1 | MTOR | 0 | 0.062 | 0.284 | 0 | 0.588 | 0.699 |
| MKNK1 | MTOR | 0 | 0.062 | 0 | 0 | 0.391 | 0.404 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, C.; Bing, Z.; Yang, L.; Huang, Z.; Wang, W.; Grebogi, C. Transcriptomic Evidence Reveals the Dysfunctional Mechanism of Synaptic Plasticity Control in ASD. Genes 2025, 16, 11. https://doi.org/10.3390/genes16010011
Kong C, Bing Z, Yang L, Huang Z, Wang W, Grebogi C. Transcriptomic Evidence Reveals the Dysfunctional Mechanism of Synaptic Plasticity Control in ASD. Genes. 2025; 16(1):11. https://doi.org/10.3390/genes16010011
Chicago/Turabian StyleKong, Chao, Zhitong Bing, Lei Yang, Zigang Huang, Wenxu Wang, and Celso Grebogi. 2025. "Transcriptomic Evidence Reveals the Dysfunctional Mechanism of Synaptic Plasticity Control in ASD" Genes 16, no. 1: 11. https://doi.org/10.3390/genes16010011
APA StyleKong, C., Bing, Z., Yang, L., Huang, Z., Wang, W., & Grebogi, C. (2025). Transcriptomic Evidence Reveals the Dysfunctional Mechanism of Synaptic Plasticity Control in ASD. Genes, 16(1), 11. https://doi.org/10.3390/genes16010011