
Screening Methods to Discover the FDA-Approved Cancer Drug Encorafenib as Optimally Selective for Metallothionein Gene Loss Ovarian Cancer
 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Line, Plasmids, Culturing Conditions, and Standard Plastics
2.2. Drug and Cadmium Sources
2.3. CellTracker™ Green Assay
2.4. CellTiter-GLO® Assay
2.5. Nuclear Localized GFP and BFP Nuclei Counting
2.6. Crystal Violet Viability Assay
2.7. Hoechst 33342 Nuclear Count Viability Assay
2.8. Drug Screen of an FDA-Approved Drug in Cancer Indication
2.9. Western Immunoblots
2.10. Morphological Brightfield Microscopy
3. Results
3.1. Genetics of Metallothioneins in Ovarian Cancer
3.2. Metallothionein Controlled Zinc and Cadmium Toxicity
3.3. Assessment of Screening Methods
3.3.1. Live Cell Methods
3.3.2. Fixed Cell Methods
3.3.3. Summary of Methods and Assay Choice
3.3.4. Robotic Method
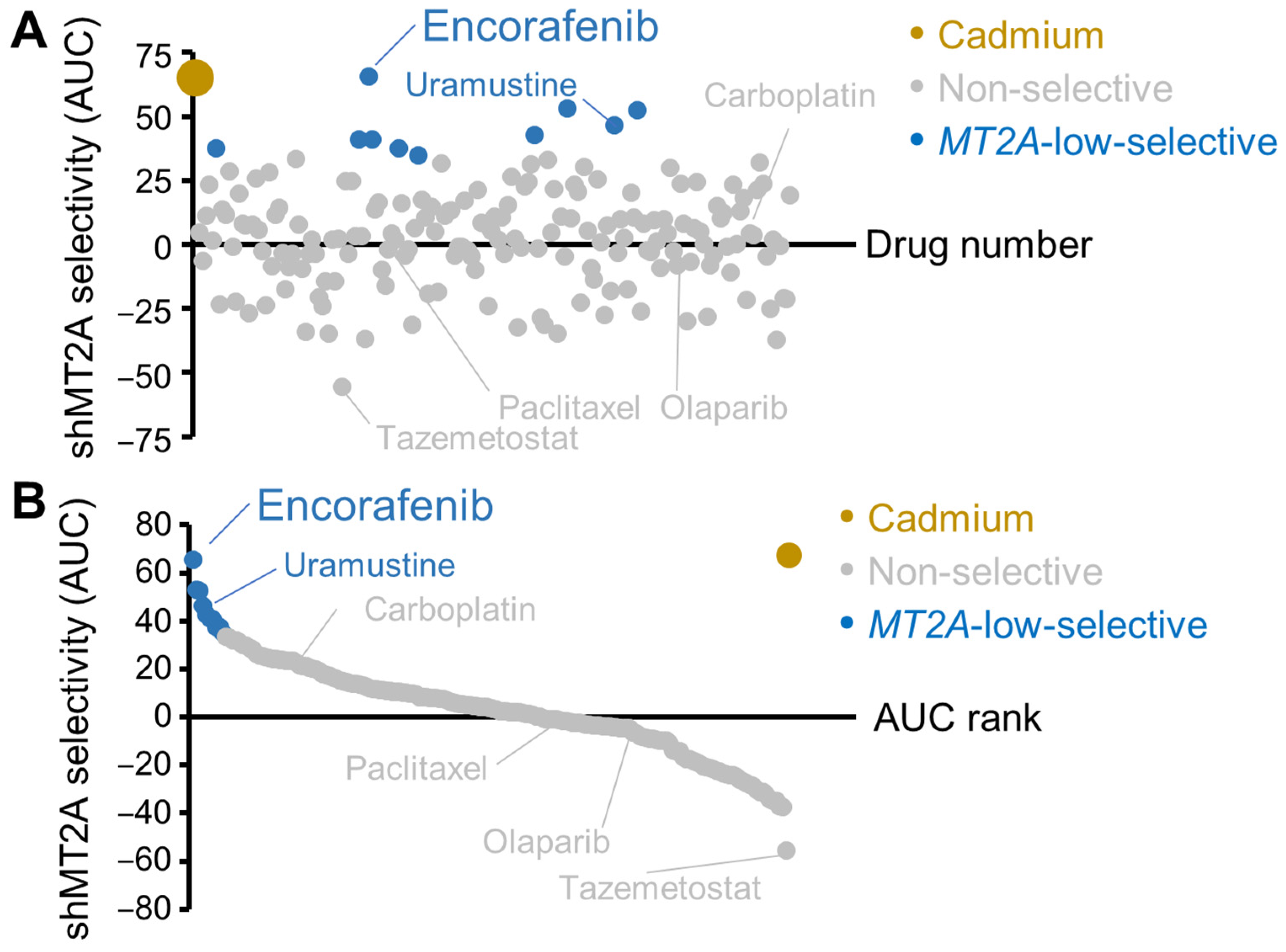
3.4. Screen of FDA-Approved Cancer Drugs for Low Metallothionein Selectivity
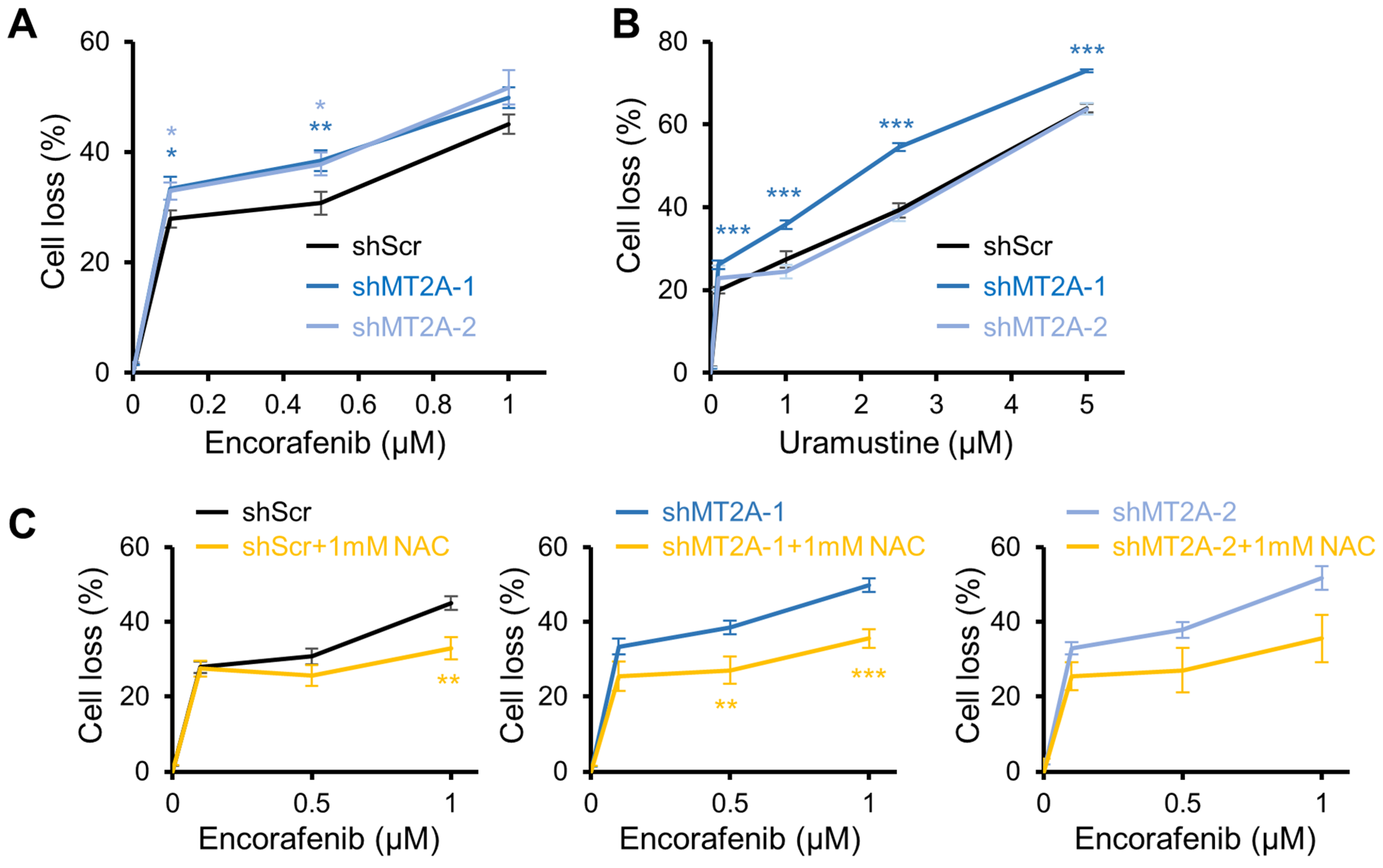
3.5. Encorafenib Selectivity Targets Engineered Low Metallothionein Ovarian Cancer
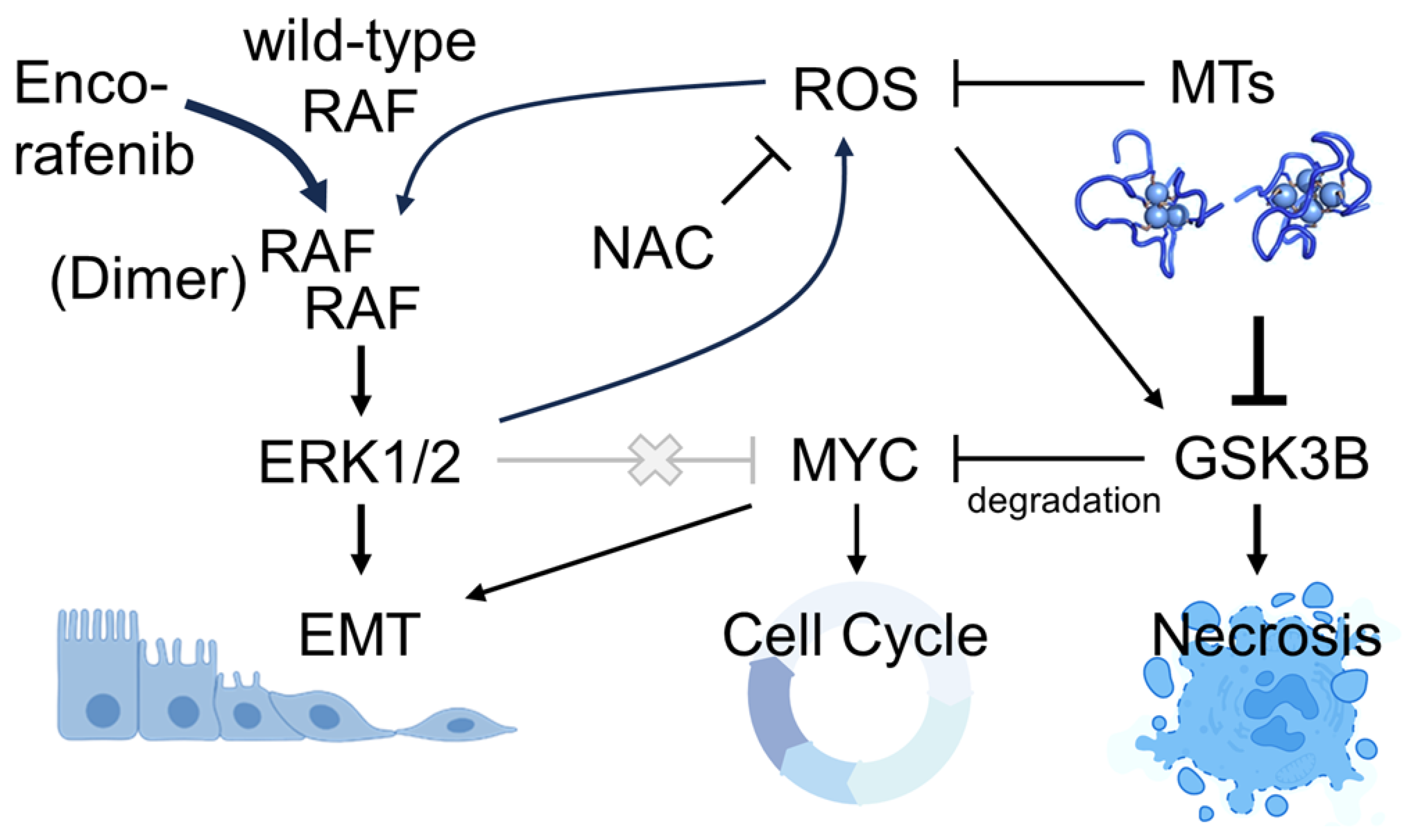
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowers, R.R.; Jones, C.M.; Paz, E.A.; Barrows, J.K.; Armeson, K.E.; Long, D.T.; Delaney, J.R. SWAN pathway-network identification of common aneuploidy-based oncogenic drivers. Nucleic Acids Res. 2022, 50, 3673–3692. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.B.; Yu, B.; Yang, V.W. p53 suppresses structural chromosome instability after mitotic arrest in human cells. Oncogene 2010, 29, 1929–1940. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Ghoshal, K.; Jacob, S.T. Regulation of metallothionein gene expression. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 66, 357–384. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thirumoorthy, N.; Shyam Sunder, A.; Manisenthil Kumar, K.; Senthil Kumar, M.; Ganesh, G.; Chatterjee, M. A review of metallothionein isoforms and their role in pathophysiology. World J. Surg. Oncol. 2011, 9, 54. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Liu, J.; Choudhuri, S. Metallothionein: An intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 267–294. [Google Scholar] [CrossRef]
- Franklin, R.B.; Costello, L.C. The important role of the apoptotic effects of zinc in the development of cancers. J. Cell. Biochem. 2009, 106, 750–757. [Google Scholar] [CrossRef]
- Namdarghanbari, M.; Wobig, W.; Krezoski, S.; Tabatabai, N.M.; Petering, D.H. Mammalian metallothionein in toxicology, cancer, and cancer chemotherapy. J. Biol. Inorg. Chem. 2011, 16, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Arseniev, A.; Schultze, P.; Worgotter, E.; Braun, W.; Wagner, G.; Vasak, M.; Kagi, J.H.; Wuthrich, K. Three-dimensional structure of rabbit liver [Cd7]metallothionein-2a in aqueous solution determined by nuclear magnetic resonance. J. Mol. Biol. 1988, 201, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. An ecological study of cancer mortality rates including indices for dietary iron and zinc. Anticancer Res. 2008, 28, 1955–1963. [Google Scholar] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yu, P.; Chan, W.N.; Xie, F.; Zhang, Y.; Liang, L.; Leung, K.T.; Lo, K.W.; Yu, J.; Tse, G.M.K.; et al. Cellular zinc metabolism and zinc signaling: From biological functions to diseases and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 6. [Google Scholar] [CrossRef]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Saydam, N.; Adams, T.K.; Steiner, F.; Schaffner, W.; Freedman, J.H. Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: Identification of signal transduction cascades that control metal-inducible transcription. J. Biol. Chem. 2002, 277, 20438–20445. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.R.; Stupack, D.G. Whole Genome Pathway Analysis Identifies an Association of Cadmium Response Gene Loss with Copy Number Variation in Mutant p53 Bearing Uterine Endometrial Carcinomas. PLoS ONE 2016, 11, e0159114. [Google Scholar] [CrossRef]
- Lei, Z.N.; Tian, Q.; Teng, Q.X.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z.S. Understanding and targeting resistance mechanisms in cancer. MedComm 2023, 4, e265. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Anticancer drug resistance: An update and perspective. Drug Resist. Updat. 2021, 59, 100796. [Google Scholar] [CrossRef]
- Delaney, J.R.; Patel, C.B.; Willis, K.M.; Haghighiabyaneh, M.; Axelrod, J.; Tancioni, I.; Lu, D.; Bapat, J.; Young, S.; Cadassou, O.; et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat. Commun. 2017, 8, 14423. [Google Scholar] [CrossRef]
- Bowers, R.R.; Andrade, M.F.; Jones, C.M.; White-Gilbertson, S.; Voelkel-Johnson, C.; Delaney, J.R. Autophagy modulating therapeutics inhibit ovarian cancer colony generation by polyploid giant cancer cells (PGCCs). BMC Cancer 2022, 22, 410. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef] [PubMed]
- Ortell, K.K.; Switonski, P.M.; Delaney, J.R. FairSubset: A tool to choose representative subsets of data for use with replicates or groups of different sample sizes. J. Biol. Methods 2019, 6, e118. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087379. [Google Scholar] [CrossRef] [PubMed]
- Chazotte, B. Labeling nuclear DNA with hoechst 33342. Cold Spring Harb. Protoc. 2011, 2011, pdb-prot5557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. USA 1994, 91, 237–241. [Google Scholar] [CrossRef]
- Jiang, L.J.; Maret, W.; Vallee, B.L. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc. Natl. Acad. Sci. USA 1998, 95, 3483–3488. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhitkovich, A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019, 32, 1318–1319. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, J.; Ippolito, M.R.; Eliezer, Y.; De Feudis, G.; Reuveni, E.; Savir Kadmon, A.; Martin, S.; Vigano, S.; Leor, G.; Berstler, J.; et al. Human aneuploid cells depend on the RAF/MEK/ERK pathway for overcoming increased DNA damage. Nat. Commun. 2024, 15, 7772. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Hussain, J.; Giblett, S.; Hayward, R.; Marais, R.; Pritchard, C. BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res. 2010, 70, 8475–8486. [Google Scholar] [CrossRef]
- Ren, L.X.; Qi, J.C.; Zhao, A.N.; Shi, B.; Zhang, H.; Wang, D.D.; Yang, Z. Myc-associated zinc-finger protein promotes clear cell renal cell carcinoma progression through transcriptional activation of the MAP2K2-dependent ERK pathway. Cancer Cell Int. 2021, 21, 323. [Google Scholar] [CrossRef]
- Wei, S.; Zhang, J.; Han, B.; Liu, J.; Xiang, X.; Zhang, M.; Xia, S.; Zhang, W.; Zhang, X. Novel Zinc Finger Transcription Factor ZFP580 Facilitates All-Trans Retinoic Acid -Induced Vascular Smooth Muscle Cells Differentiation by Raralpha-Mediated PI3K/Akt and ERK Signaling. Cell. Physiol. Biochem. 2018, 50, 2390–2405. [Google Scholar] [CrossRef]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. Cadmium and cancer. Met. Ions Life Sci. 2013, 11, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, L.L.; Gonzalez-Navajas, J.; Seo, G.S.; Shen, C.; Brick, J.; Herdman, S.; Varki, N.; Corr, M.; Lee, J.; et al. ERK activation drives intestinal tumorigenesis in Apcmin/+ mice. Nat. Med. 2010, 16, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Ciotti, S.; Iuliano, L.; Cefalu, S.; Comelli, M.; Mavelli, I.; Di Giorgio, E.; Brancolini, C. GSK3beta is a key regulator of the ROS-dependent necrotic death induced by the quinone DMNQ. Cell Death Dis. 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Miller, A.; Brady, W.E.; Paul, J.; Carty, K.; Rodgers, W.; Millan, D.; Coleman, R.L.; Moore, K.N.; Banerjee, S.; et al. Trametinib versus standard of care in patients with recurrent low-grade serous ovarian cancer (GOG 281/LOGS): An international, randomised, open-label, multicentre, phase 2/3 trial. Lancet 2022, 399, 541–553. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Value Rank | Standard Deviation (%) | Strengths | Limitations |
|---|---|---|---|---|
| Hoechst 33342 (fixed) | 1 | 3.2 |
|
|
| nls-GFP, counts (live) | 2 | 3.9 |
|
|
| nls-GFP/BFP, competition (live) | 3 | 13.9 |
|
|
| Crystal violet (fixed) | 4 | 4.3 * |
|
|
| CellTiter-GLO® (live) | 5 | 7.9 |
|
|
| CellTracker™ Green (live) | 6 | 25.6 |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rees, A.; Villamor, E.; Evans, D.; Gooz, M.; Fallon, C.; Mina-Abouda, M.; Disharoon, A.; Eblen, S.T.; Delaney, J.R. Screening Methods to Discover the FDA-Approved Cancer Drug Encorafenib as Optimally Selective for Metallothionein Gene Loss Ovarian Cancer. Genes 2025, 16, 42. https://doi.org/10.3390/genes16010042
Rees A, Villamor E, Evans D, Gooz M, Fallon C, Mina-Abouda M, Disharoon A, Eblen ST, Delaney JR. Screening Methods to Discover the FDA-Approved Cancer Drug Encorafenib as Optimally Selective for Metallothionein Gene Loss Ovarian Cancer. Genes. 2025; 16(1):42. https://doi.org/10.3390/genes16010042
Chicago/Turabian StyleRees, Amy, Evan Villamor, Della Evans, Monika Gooz, Clare Fallon, Mirna Mina-Abouda, Andrew Disharoon, Scott T. Eblen, and Joe R. Delaney. 2025. "Screening Methods to Discover the FDA-Approved Cancer Drug Encorafenib as Optimally Selective for Metallothionein Gene Loss Ovarian Cancer" Genes 16, no. 1: 42. https://doi.org/10.3390/genes16010042
APA StyleRees, A., Villamor, E., Evans, D., Gooz, M., Fallon, C., Mina-Abouda, M., Disharoon, A., Eblen, S. T., & Delaney, J. R. (2025). Screening Methods to Discover the FDA-Approved Cancer Drug Encorafenib as Optimally Selective for Metallothionein Gene Loss Ovarian Cancer. Genes, 16(1), 42. https://doi.org/10.3390/genes16010042