
Lipoprotein Lipase: Structure, Function, and Genetic Variation

Highlights
- An up-to-date curated list of 300 LPL gene variants in patients with hypertriglyceridemia, specifically familial chylomicronemia syndrome (FCS).
- Analysis and discussion of LPL pathogenic variants and other variant types.
- An overview of LPL expression, regulation, structure, and physiological roles.
- Our variant list is a unique resource for those interested in the clinical relevance of LPL variants related to FCS.
- To our knowledge, this is the most complete exon-by-exon curation of LPL genetic variants and can serve as a bridge towards the future development of a public database of disease-associated variants.
- The variants are interpreted in the context of current understanding of the biology and biochemistry of LPL.
Abstract
:1. Introduction
2. Synthesis and Expression of LPL
3. Physiological Roles of LPL in Lipoprotein Metabolism and Their Regulation
4. Protein Structure of LPL
5. Genomic Structure of LPL
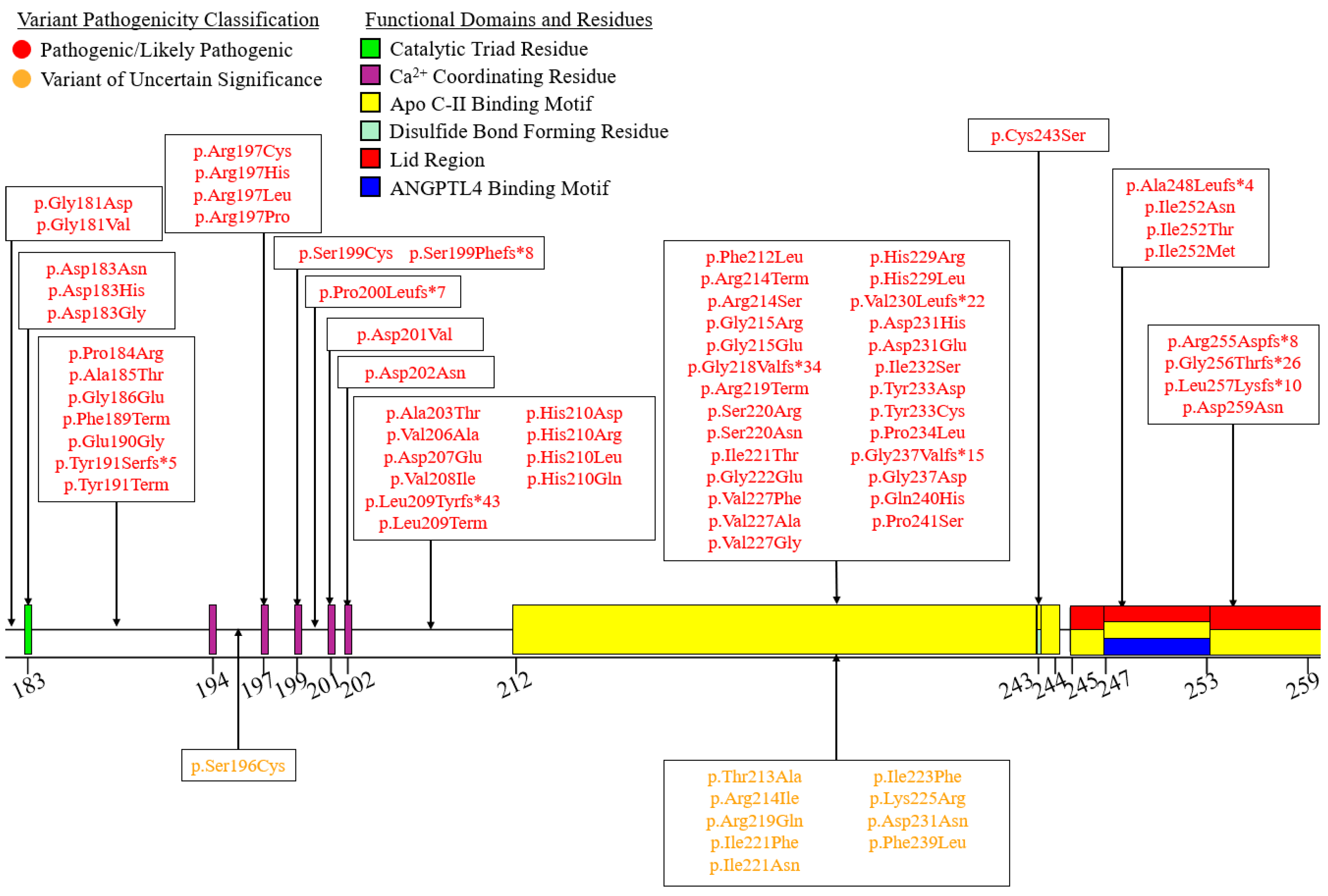
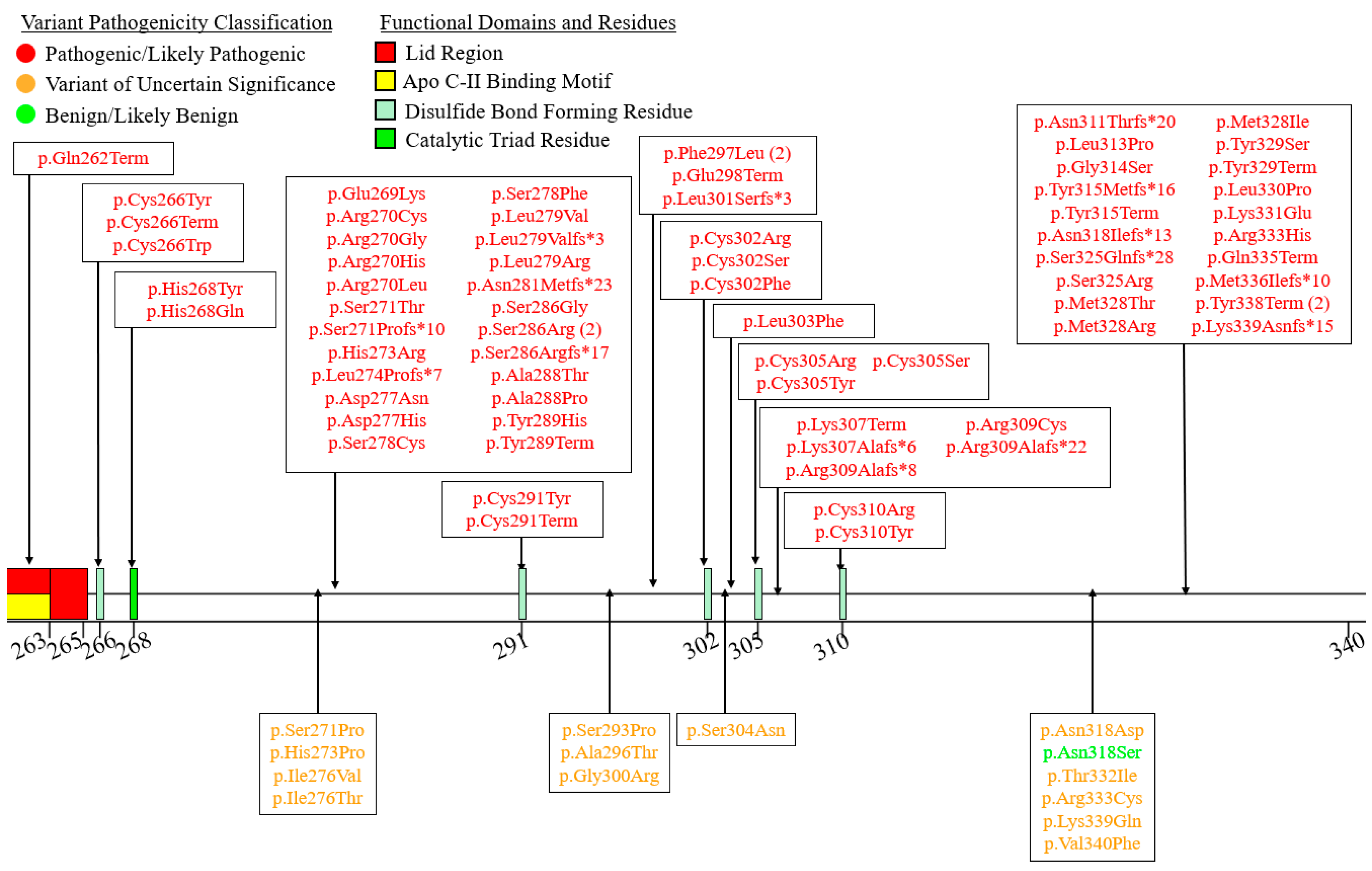
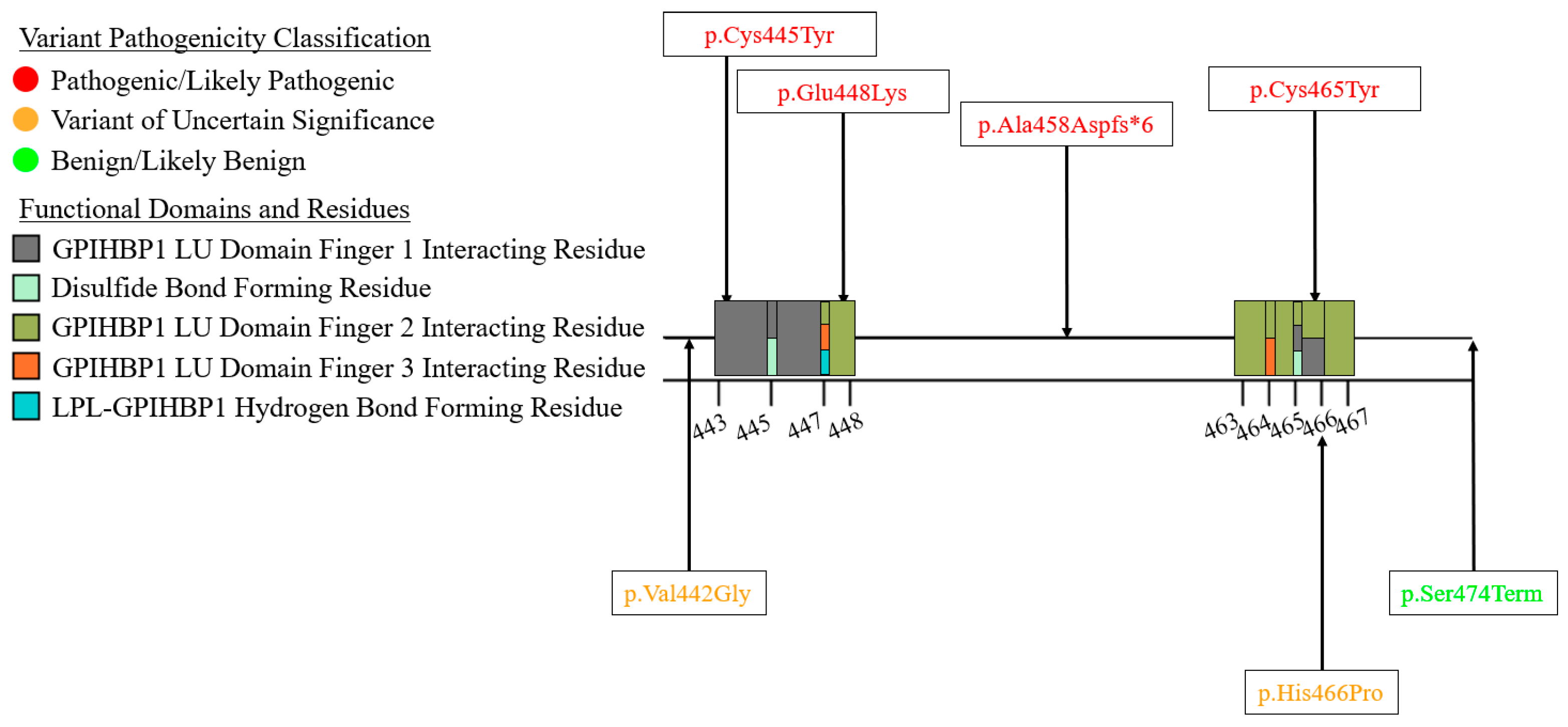
6. A Curated Assembly of LPL Variants
7. Coding Region Variants
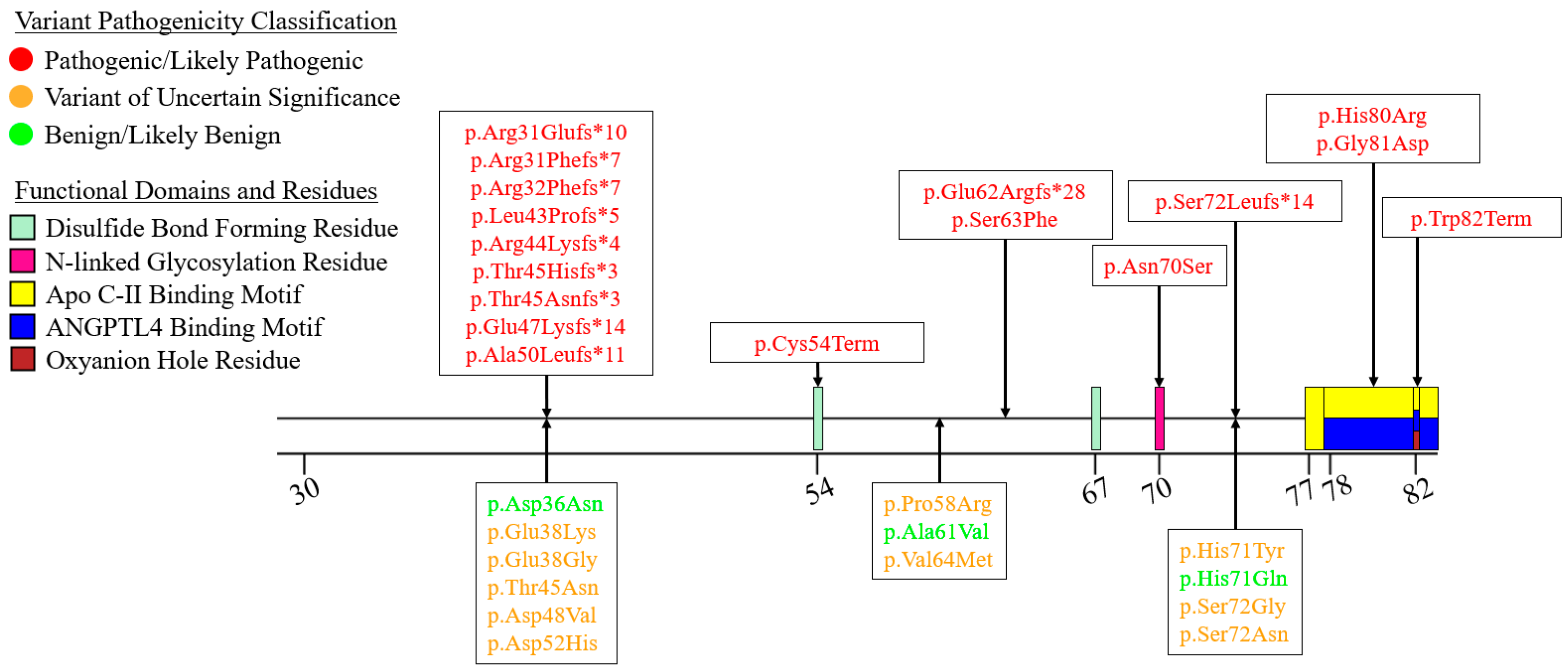
7.1. Exon 1—Signal Peptide
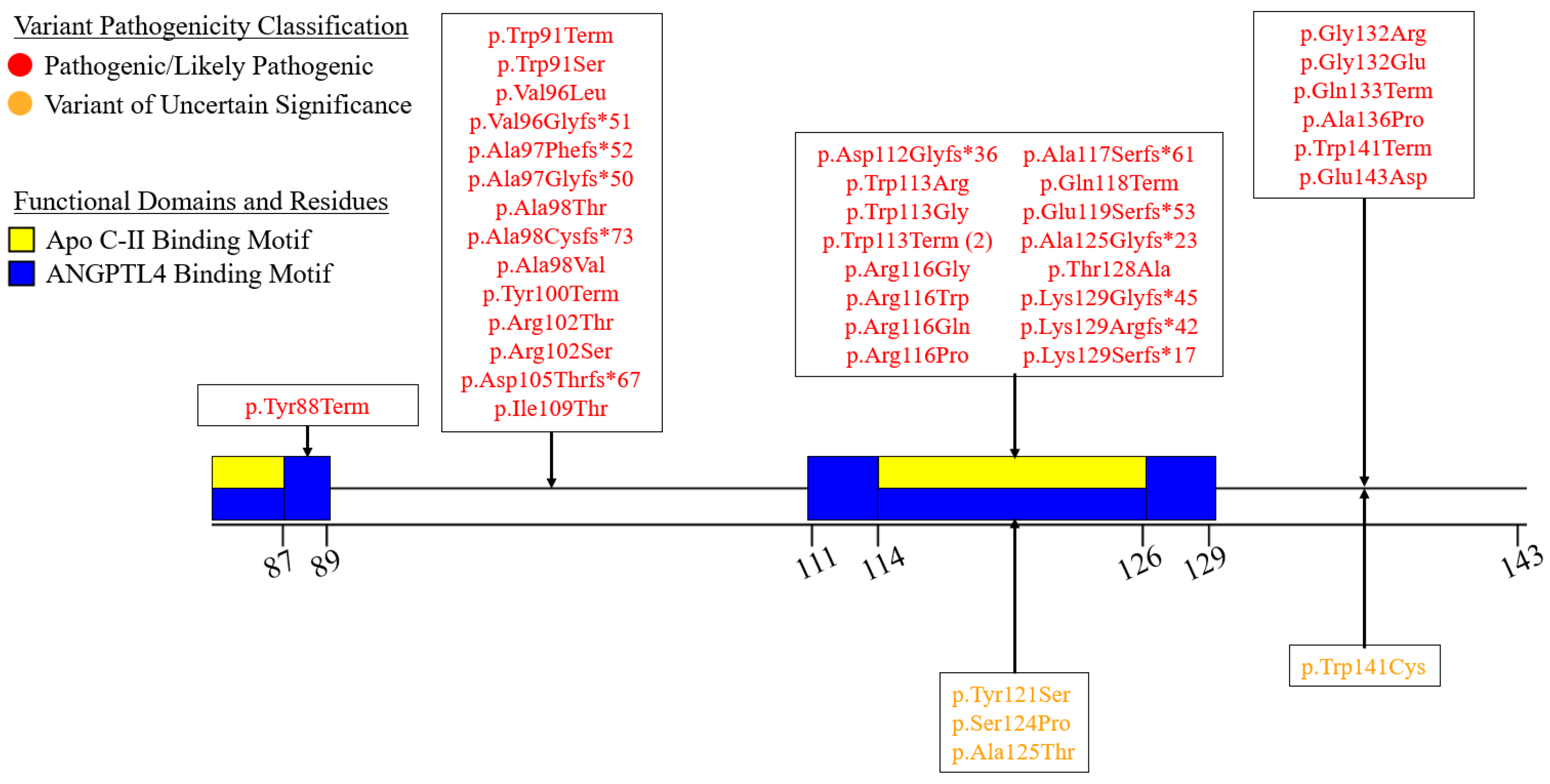
7.2. Exons 2–6—N-Terminal α/β-Hydrolase Domain
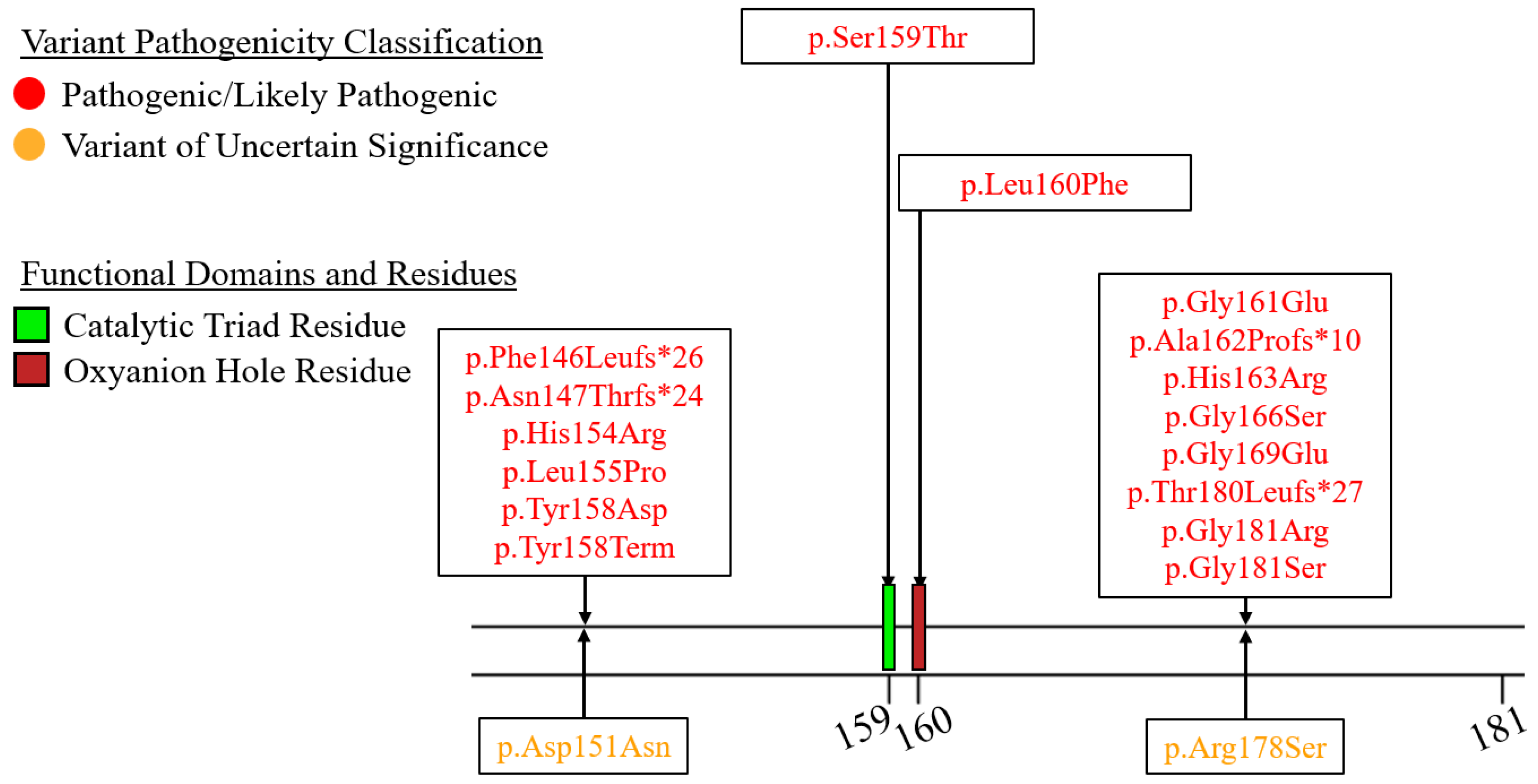
7.3. Exons 7–9—C-Terminal Lipid Binding Domain
8. Non-Coding and Large-Scale Variants
9. Benign/Likely Benign Variants: Deleterious and Gain-of-Function Variants
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, S.G.; Zechner, R. Biochemistry and Pathophysiology of Intravascular and Intracellular Lipolysis. Genes. Dev. 2013, 27, 459–484. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Berberich, A.J.; Ban, M.R.; Wang, J.; Digenio, A.; Alexander, V.J.; D’Erasmo, L.; Arca, M.; Jones, A.; Bruckert, E.; et al. Clinical and Biochemical Features of Different Molecular Etiologies of Familial Chylomicronemia. J. Clin. Lipidol. 2018, 12, 920–927.e4. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Wang, J.; McIntyre, A.D.; Dron, J.S.; Hegele, R.A. The Longitudinal Triglyceride Phenotype in Heterozygotes with LPL Pathogenic Variants. J. Clin. Lipidol. 2023, 17, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Physiological Regulation of Lipoprotein Lipase. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2014, 1841, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Goulbourne, C.N.; Gin, P.; Tatar, A.; Nobumori, C.; Hoenger, A.; Jiang, H.; Grovenor, C.R.M.; Adeyo, O.; Esko, J.D.; Goldberg, I.J.; et al. The GPIHBP1–LPL Complex Is Responsible for the Margination of Triglyceride-Rich Lipoproteins in Capillaries. Cell Metab. 2014, 19, 849–860. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Ben-Zeev, O.; Stahnke, G.; Liu, G.; Davis, R.C.; Doolittle, M.H. Lipoprotein Lipase and Hepatic Lipase: The Role of Asparagine-Linked Glycosylation in the Expression of a Functional Enzyme. J. Lipid Res. 1994, 35, 1511–1523. [Google Scholar] [CrossRef]
- Lo, J.Y.; Smith, L.C.; Chan, L. Lipoprotein Lipase: Role of Intramolecular Disulfide Bonds in Enzyme Catalysis. Biochem. Biophys. Res. Commun. 1995, 206, 266–271. [Google Scholar] [CrossRef]
- Roberts, B.S.; Babilonia-Rosa, M.A.; Broadwell, L.J.; Wu, M.J.; Neher, S.B. Lipase Maturation Factor 1 Affects Redox Homeostasis in the Endoplasmic Reticulum. EMBO J. 2018, 37, e97379. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR Pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Sha, H.; Sun, S.; Francisco, A.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.; Panhwar, M.S.; et al. The ER-Associated Degradation Adaptor Protein Sel1L Regulates LPL Secretion and Lipid Metabolism. Cell Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Ailhaud, G. Cellular and Secreted Lipoprotein Lipase Revisited. Clin. Biochem. 1990, 23, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, E.L.; Deng, Y.; Burd, C.G. Syndecan-1 Mediates Sorting of Soluble Lipoprotein Lipase with Sphingomyelin-Rich Membrane in the Golgi Apparatus. Dev. Cell 2019, 51, 387–398.e4. [Google Scholar] [CrossRef]
- Klinger, S.C.; Glerup, S.; Raarup, M.K.; Mari, M.C.; Nyegaard, M.; Koster, G.; Prabakaran, T.; Nilsson, S.K.; Kjaergaard, M.M.; Bakke, O.; et al. SorLA Regulates the Activity of Lipoprotein Lipase by Intracellular Trafficking. J. Cell Sci. 2011, 124, 1095–1105. [Google Scholar] [CrossRef]
- Bankaitis, V.A.; Wang, Y. Lipoprotein Lipase Sorting: Sphingomyelin and a Proteoglycan Show the Way. Trends Cell Biol. 2020, 30, 170–172. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Rothman, J.E. Biosynthetic protein transport and sorting by the endoplasmic reticulum and golgi. Annu. Rev. Biochem. 1987, 56, 829–852. [Google Scholar] [CrossRef]
- Gunn, K.H.; Roberts, B.S.; Wang, F.; Strauss, J.D.; Borgnia, M.J.; Egelman, E.H.; Neher, S.B. The Structure of Helical Lipoprotein Lipase Reveals an Unexpected Twist in Lipase Storage. Proc. Natl. Acad. Sci. USA 2020, 117, 10254–10264. [Google Scholar] [CrossRef]
- Allan, C.M.; Larsson, M.; Jung, R.S.; Ploug, M.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; Young, S.G. Mobility of “HSPG-Bound” LPL Explains How LPL Is Able to Reach GPIHBP1 on Capillaries. J. Lipid Res. 2017, 58, 216–225. [Google Scholar] [CrossRef]
- Davies, B.S.J.; Beigneux, A.P.; Barnes, R.H.; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyrén, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 Is Responsible for the Entry of Lipoprotein Lipase into Capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef]
- Young, S.G.; Fong, L.G.; Beigneux, A.P.; Allan, C.M.; He, C.; Jiang, H.; Nakajima, K.; Meiyappan, M.; Birrane, G.; Ploug, M. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019, 30, 51–65. [Google Scholar] [CrossRef]
- Davies, B.S.J.; Goulbourne, C.N.; Barnes, R.H.; Turlo, K.A.; Gin, P.; Vaughan, S.; Vaux, D.J.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; et al. Assessing Mechanisms of GPIHBP1 and Lipoprotein Lipase Movement across Endothelial Cells. J. Lipid Res. 2012, 53, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.F. Abolishment of Alimentary Lipemia Following Injection of Heparin. Science 1943, 98, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.D. Clearing Factor, a Heparin-Activated Lipoprotein Lipase. I. Isolation and Characterization of the Enzyme from Normal Rat Heart. J. Biol. Chem. 1955, 215, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.D. Clearing Factor, a Heparin-Activated Lipoprotein Lipase. II. Substrate Specificity and Activation of Coconut Oil. J. Biol. Chem. 1955, 215, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein Lipase: Structure, Function, Regulation, and Role in Disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Brewer, H.B. Lipoprotein Lipase: Structure, Function and Mechanism of Action. Int. J. Clin. Lab. Res. 1994, 24, 143–147. [Google Scholar] [CrossRef]
- Birrane, G.; Beigneux, A.P.; Dwyer, B.; Strack-Logue, B.; Kristensen, K.K.; Francone, O.L.; Fong, L.G.; Mertens, H.D.T.; Pan, C.Q.; Ploug, M.; et al. Structure of the Lipoprotein Lipase–GPIHBP1 Complex That Mediates Plasma Triglyceride Hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 1723–1732. [Google Scholar] [CrossRef]
- Arora, R.; Nimonkar, A.V.; Baird, D.; Wang, C.; Chiu, C.-H.; Horton, P.A.; Hanrahan, S.; Cubbon, R.; Weldon, S.; Tschantz, W.R.; et al. Structure of Lipoprotein Lipase in Complex with GPIHBP1. Proc. Natl. Acad. Sci. USA 2019, 116, 10360–10365. [Google Scholar] [CrossRef]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New Findings Related to Genetics, Biochemistry, and Role in Triglyceride Metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef]
- Kumari, A.; Grønnemose, A.L.; Kristensen, K.K.; Winther, A.-M.L.; Young, S.G.; Jørgensen, T.J.D.; Ploug, M. Inverse Effects of APOC2 and ANGPTL4 on the Conformational Dynamics of Lid-Anchoring Structures in Lipoprotein Lipase. Proc. Natl. Acad. Sci. USA 2023, 120, e2221888120. [Google Scholar] [CrossRef]
- Gin, P.; Yin, L.; Davies, B.S.J.; Weinstein, M.M.; Ryan, R.O.; Bensadoun, A.; Fong, L.G.; Young, S.G.; Beigneux, A.P. The Acidic Domain of GPIHBP1 Is Important for the Binding of Lipoprotein Lipase and Chylomicrons∗. J. Biol. Chem. 2008, 283, 29554–29562. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Nelbach, L.; Weinstein, M.M.; Burgess, B.L.; Beckstead, J.A.; Young, S.G.; Ryan, R.O.; Forte, T.M. Intravenous Injection of apoA-V Reconstituted HDL Decreases Hypertriglyceridemia in Apoav−/− Mice and Requires GPIHBP1. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Hegele, R.A. Genetic Variation in Apolipoprotein A-V in Hypertriglyceridemia. Curr. Opin. Lipidol. 2024, 35, 66. [Google Scholar] [CrossRef]
- Shetty, S.K.; Walzem, R.L.; Davies, B.S.J. A Novel NanoBiT-Based Assay Monitors the Interaction between Lipoprotein Lipase and GPIHBP1 in Real Time[S]. J. Lipid Res. 2020, 61, 546–559. [Google Scholar] [CrossRef]
- Gunn, K.H.; Neher, S.B. Structure of Dimeric Lipoprotein Lipase Reveals a Pore Adjacent to the Active Site. Nat. Commun. 2023, 14, 2569. [Google Scholar] [CrossRef]
- Zambon, A.; Schmidt, I.; Beisiegel, U.; Brunzell, J.D. Dimeric Lipoprotein Lipase Is Bound to Triglyceride-Rich Plasma Lipoproteins. J. Lipid Res. 1996, 37, 2394–2404. [Google Scholar] [CrossRef]
- Krapp, A.; Zhang, H.; Ginzinger, D.; Liu, M.S.; Lindberg, A.; Olivecrona, G.; Hayden, M.R.; Beisiegel, U. Structural Features in Lipoprotein Lipase Necessary for the Mediation of Lipoprotein Uptake into Cells. J. Lipid Res. 1995, 36, 2362–2373. [Google Scholar] [CrossRef]
- Nykjaer, A.; Bengtsson-Olivecrona, G.; Lookene, A.; Moestrup, S.K.; Petersen, C.M.; Weber, W.; Beisiegel, U.; Gliemann, J. The α2-Macroglobulin Receptor/Low Density Lipoprotein Receptor-Related Protein Binds Lipoprotein Lipase and β-Migrating Very Low Density Lipoprotein Associated with the Lipase. J. Biol. Chem. 1993, 268, 15048–15055. [Google Scholar] [CrossRef]
- Mamputu, J.C.; Desfaits, A.C.; Renier, G. Lipoprotein Lipase Enhances Human Monocyte Adhesion to Aortic Endothelial Cells. J. Lipid Res. 1997, 38, 1722–1729. [Google Scholar] [CrossRef]
- Mamputu, J.-C.; Levesque, L.; Renier, G. Proliferative Effect of Lipoprotein Lipase on Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2212–2219. [Google Scholar] [CrossRef]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J.; et al. Integrative Genomic Analysis Implicates Limited Peripheral Adipose Storage Capacity in the Pathogenesis of Human Insulin Resistance. Nat. Genet. 2017, 49, 17–26. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; O’Dushlaine, C.; Liu, D.; Stitziel, N.O.; Natarajan, P.; Nomura, A.; Emdin, C.A.; Gupta, N.; et al. Association of Rare and Common Variation in the Lipoprotein Lipase Gene with Coronary Artery Disease. JAMA 2017, 317, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Han, T.; Stachura, D.L.; Wang, H.; Vaisman, B.L.; Kim, J.; Klemke, R.L.; Remaley, A.T.; Rana, T.M.; Traver, D.; et al. Lipoprotein Lipase Regulates Hematopoietic Stem Progenitor Cell Maintenance through DHA Supply. Nat. Commun. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, Y.Q.; Konrad, R.J. The Regulation of Triacylglycerol Metabolism and Lipoprotein Lipase Activity. Adv. Biol. 2022, 6, 2200093. [Google Scholar] [CrossRef]
- Oldoni, F.; Cheng, H.; Banfi, S.; Gusarova, V.; Cohen, J.C.; Hobbs, H.H. ANGPTL8 Has Both Endocrine and Autocrine Effects on Substrate Utilization. JCI Insight 2020, 5, e138777. [Google Scholar] [CrossRef]
- Sylvers-Davie, K.L.; Davies, B.S.J. Regulation of Lipoprotein Metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E493–E508. [Google Scholar] [CrossRef]
- Chi, X.; Britt, E.C.; Shows, H.W.; Hjelmaas, A.J.; Shetty, S.K.; Cushing, E.M.; Li, W.; Dou, A.; Zhang, R.; Davies, B.S.J. ANGPTL8 Promotes the Ability of ANGPTL3 to Bind and Inhibit Lipoprotein Lipase. Mol. Metab. 2017, 6, 1137–1149. [Google Scholar] [CrossRef]
- Su, X.; Kong, Y.; Peng, D. New Insights into Apolipoprotein A5 in Controlling Lipoprotein Metabolism in Obesity and the Metabolic Syndrome Patients. Lipids Health Dis. 2018, 17, 174. [Google Scholar] [CrossRef]
- Wolska, A.; Reimund, M.; Remaley, A.T. Apolipoprotein C-II: The Re-Emergence of a Forgotten Factor. Curr. Opin. Lipidol. 2020, 31, 147. [Google Scholar] [CrossRef]
- Ramms, B.; Gordts, P.L.S.M. Apolipoprotein C-III in Triglyceride-Rich Lipoprotein Metabolism. Curr. Opin. Lipidol. 2018, 29, 171. [Google Scholar] [CrossRef] [PubMed]
- Berbée, J.F.P.; Hoogt, C.C.v.d.; Sundararaman, D.; Havekes, L.M.; Rensen, P.C.N. Severe Hypertriglyceridemia in Human APOC1 Transgenic Mice Is Caused by apoC-I-Induced Inhibition of LPL. J. Lipid Res. 2005, 46, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Haan, W.d.; Berbeée, J.F.P.; Havekes, L.M.; Rensen, P.C.N. Endogenous apoC-I Increases Hyperlipidemia in apoE-Knockout Mice by Stimulating VLDL Production and Inhibiting LPL. J. Lipid Res. 2006, 47, 1203–1211. [Google Scholar] [CrossRef]
- Larsson, M.; Vorrsjö, E.; Talmud, P.; Lookene, A.; Olivecrona, G. Apolipoproteins C-I and C-III Inhibit Lipoprotein Lipase Activity by Displacement of the Enzyme from Lipid Droplets. J. Biol. Chem. 2013, 288, 33997–34008. [Google Scholar] [CrossRef]
- Rensen, P.C.N.; Berkel, T.J.C. van Apolipoprotein E Effectively Inhibits Lipoprotein Lipase-Mediated Lipolysis of Chylomicron-like Triglyceride-Rich Lipid Emulsions in Vitro and in Vivo*. J. Biol. Chem. 1996, 271, 14791–14799. [Google Scholar] [CrossRef]
- Phillips, M.C. Apolipoprotein E Isoforms and Lipoprotein Metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure Determines Function, from Atherosclerosis to Alzheimer’s Disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef]
- Balasubramaniam, D.; Schroeder, O.; Russell, A.M.; Fitchett, J.R.; Austin, A.K.; Beyer, T.P.; Chen, Y.Q.; Day, J.W.; Ehsani, M.; Heng, A.R.; et al. An Anti-ANGPTL3/8 Antibody Decreases Circulating Triglycerides by Binding to a LPL-Inhibitory Leucine Zipper-like Motif. J. Lipid Res. 2022, 63, 100198. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Konrad, R.J. ApoA5 Lowers Triglyceride Levels via Suppression of ANGPTL3/8-Mediated LPL Inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, K. An Updated ANGPTL3-4-8 Model as a Mechanism of Triglyceride Partitioning between Fat and Oxidative Tissues. Progress. Lipid Res. 2022, 85, 101140. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Zhen, E.Y.; Regmi, A.; Roell, W.C.; Guo, H.; Luo, M.J.; et al. Angiopoietin-like Protein 8 Differentially Regulates ANGPTL3 and ANGPTL4 during Postprandial Partitioning of Fatty Acids. J. Lipid Res. 2020, 61, 1203–1220. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Allan, C.M.; Jung, R.S.; Heizer, P.J.; Beigneux, A.P.; Young, S.G.; Fong, L.G. Apolipoprotein C-III Inhibits Triglyceride Hydrolysis by GPIHBP1-Bound LPL. J. Lipid Res. 2017, 58, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, B.E.; Howles, P.; Street, S.; Morris, J.; Swertfeger, D.; Davidson, W.S. Apolipoprotein E Content of VLDL Limits LPL-Mediated Triglyceride Hydrolysis. J. Lipid Res. 2022, 63, 100157. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.F.; Mintah, I.J.; Shihanian, L.M.; Stevis, P.; Buckler, D.; Alexa-Braun, C.A.; Kleiner, S.; Banfi, S.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL8 Requires ANGPTL3 to Inhibit Lipoprotein Lipase and Plasma Triglyceride Clearance. J. Lipid Res. 2017, 58, 1166–1173. [Google Scholar] [CrossRef]
- Kovrov, O.; Kristensen, K.K.; Larsson, E.; Ploug, M.; Olivecrona, G. On the Mechanism of Angiopoietin-like Protein 8 for Control of Lipoprotein Lipase Activity. J. Lipid Res. 2019, 60, 783–793. [Google Scholar] [CrossRef]
- Loeffler, B.; Heeren, J.; Blaeser, M.; Radner, H.; Kayser, D.; Aydin, B.; Merkel, M. Lipoprotein Lipase-Facilitated Uptake of LDL Is Mediated by the LDL Receptor. J. Lipid Res. 2007, 48, 288–298. [Google Scholar] [CrossRef]
- Rinninger, F.; Kaiser, T.; Mann, W.A.; Meyer, N.; Greten, H.; Beisiegel, U. Lipoprotein Lipase Mediates an Increase in the Selective Uptake of High Density Lipoprotein-Associated Cholesteryl Esters by Hepatic Cells in Culture. J. Lipid Res. 1998, 39, 1335–1348. [Google Scholar] [CrossRef]
- Beisiegel, U.; Weber, W.; Bengtsson-Olivecrona, G. Lipoprotein Lipase Enhances the Binding of Chylomicrons to Low Density Lipoprotein Receptor-Related Protein. Proc. Natl. Acad. Sci. USA 1991, 88, 8342–8346. [Google Scholar] [CrossRef]
- Chappell, D.A.; Fry, G.L.; Waknitz, M.A.; Muhonen, L.E.; Pladet, M.W.; Iverius, P.H.; Strickland, D.K. Lipoprotein Lipase Induces Catabolism of Normal Triglyceride-Rich Lipoproteins via the Low Density Lipoprotein Receptor-Related Protein/α2-Macroglobulin Receptor in Vitro. A Process Facilitated by Cell-Surface Proteoglycans. J. Biol. Chem. 1993, 268, 14168–14175. [Google Scholar] [CrossRef]
- Kounnas, M.Z.; Chappell, D.A.; Strickland, D.K.; Argraves, W.S. Glycoprotein 330, a Member of the Low Density Lipoprotein Receptor Family, Binds Lipoprotein Lipase in Vitro. J. Biol. Chem. 1993, 268, 14176–14181. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Martens, Y.A.; Bu, G. Lipoproteins in the Central Nervous System: From Biology to Pathobiology. Annu. Rev. Biochem. 2022, 91, 731–759. [Google Scholar] [CrossRef] [PubMed]
- Cruciani-Guglielmacci, C.; Magnan, C. Brain Lipoprotein Lipase as a Regulator of Energy Balance. Biochimie 2017, 143, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Beigneux, A.P.; Gårdsvoll, H.; Fong, L.G.; Bensadouen, A.; Jørgensen, T.J.; Young, S.G.; Ploug, M. The Acidic Domain of the Endothelial Membrane Protein GPIHBP1 Stabilizes Lipoprotein Lipase Activity by Preventing Unfolding of Its Catalytic Domain. eLife 2016, 5, e12095. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nakajima, T.; Inoue, I. Molecular Modeling of the Dimeric Structure of Human Lipoprotein Lipase and Functional Studies of the Carboxyl-Terminal Domain. Eur. J. Biochem. 2002, 269, 4701–4710. [Google Scholar] [CrossRef]
- Winkler, F.K.; D’Arcy, A.; Hunziker, W. Structure of Human Pancreatic Lipase. Nature 1990, 343, 771–774. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Allan, C.M.; Sandoval, N.P.; Cho, G.W.; Heizer, P.J.; Jung, R.S.; Stanhope, K.L.; Havel, P.J.; Birrane, G.; Meiyappan, M.; et al. Lipoprotein Lipase Is Active as a Monomer. Proc. Natl. Acad. Sci. USA 2019, 116, 6319–6328. [Google Scholar] [CrossRef]
- van Tilbeurgh, H.; Roussel, A.; Lalouel, J.M.; Cambillau, C. Lipoprotein Lipase. Molecular Model Based on the Pancreatic Lipase x-Ray Structure: Consequences for Heparin Binding and Catalysis. J. Biol. Chem. 1994, 269, 4626–4633. [Google Scholar] [CrossRef]
- Leth-Espensen, K.Z.; Kristensen, K.K.; Kumari, A.; Winther, A.-M.L.; Young, S.G.; Jørgensen, T.J.D.; Ploug, M. The Intrinsic Instability of the Hydrolase Domain of Lipoprotein Lipase Facilitates Its Inactivation by ANGPTL4-Catalyzed Unfolding. Proc. Natl. Acad. Sci. USA 2021, 118, e2026650118. [Google Scholar] [CrossRef]
- Lookene, A.; Groot, N.B.; Kastelein, J.J.P.; Olivecrona, G.; Bruin, T. Mutation of Tryptophan Residues in Lipoprotein Lipase: Effects on stability, immunoreactivity, and catalytic properties. J. Biol. Chem. 1997, 272, 766–772. [Google Scholar] [CrossRef]
- Xu, X.; Gårdsvoll, H.; Yuan, C.; Lin, L.; Ploug, M.; Huang, M. Crystal Structure of the Urokinase Receptor in a Ligand-Free Form. J. Mol. Biol. 2012, 416, 629–641. [Google Scholar] [CrossRef]
- Huang, Y.; Fedarovich, A.; Tomlinson, S.; Davies, C. Crystal Structure of CD59: Implications for Molecular Recognition of the Complement Proteins C8 and C9 in the Membrane-Attack Complex. Acta Cryst. D 2007, 63, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Gin, P.; Beigneux, A.P.; Voss, C.; Davies, B.S.J.; Beckstead, J.A.; Ryan, R.O.; Bensadoun, A.; Fong, L.G.; Young, S.G. Binding Preferences for GPIHBP1, a GPI-Anchored Protein of Capillary Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, M.H.; Ehrhardt, N.; Péterfy, M. Lipase Maturation Factor 1 (Lmf1): Structure and Role in Lipase Folding and Assembly. Curr. Opin. Lipidol. 2010, 21, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Lookene, A.; Zhang, L.; Hultin, M.; Olivecrona, G. Rapid Subunit Exchange in Dimeric Lipoprotein Lipase and Properties of the Inactive Monomer. J. Biol. Chem. 2004, 279, 49964–49972. [Google Scholar] [CrossRef]
- Osborne, J.C., Jr.; Bengtsson-Olivecrona, G.; Lee, N.S.; Olivecrona, T. Studies on Inactivation of Lipoprotein Lipase: Role of the Dimer to Monomer Dissociation. Biochemistry 1985, 24, 5606–5611. [Google Scholar] [CrossRef]
- Reimund, M.; Kovrov, O.; Olivecrona, G.; Lookene, A. Lipoprotein Lipase Activity and Interactions Studied in Human Plasma by Isothermal Titration Calorimetry. J. Lipid Res. 2017, 58, 279–288. [Google Scholar] [CrossRef]
- Deeb, S.S.; Peng, R. Structure of the Human Lipoprotein Lipase Gene. Biochemistry 1989, 28, 4131–4135. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Human. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Variability of Longitudinal Triglyceride Phenotype in Patients Heterozygous for Pathogenic APOA5 Variants. J. Clin. Lipidol. 2023, 17, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Dron, J.S.; Wang, J.; McIntyre, A.D.; Iacocca, M.A.; Robinson, J.F.; Ban, M.R.; Cao, H.; Hegele, R.A. Six Years’ Experience with LipidSeq: Clinical and Research Learnings from a Hybrid, Targeted Sequencing Panel for Dyslipidemias. BMC Med. Genom. 2020, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Deshotels, M.R.; Hadley, T.D.; Roth, M.; Agha, A.M.; Pulipati, V.P.; Nugent, A.K.; Virani, S.S.; Nambi, V.; Moriarty, P.M.; Davidson, M.H.; et al. Genetic Testing for Hypertriglyceridemia in Academic Lipid Clinics: Implications for Precision Medicine-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1461–1467. [Google Scholar] [CrossRef]
- Laurie, A.D.; Kyle, C.V. A Novel Frameshift Mutation in the Lipoprotein Lipase Gene Is Rescued by Alternative Messenger RNA Splicing. J. Clin. Lipidol. 2017, 11, 357–361. [Google Scholar] [CrossRef]
- Okubo, M.; Horinishi, A.; Saito, M.; Ebara, T.; Endo, Y.; Kaku, K.; Murase, T.; Eto, M. A Novel Complex Deletion–Insertion Mutation Mediated by Alu Repetitive Elements Leads to Lipoprotein Lipase Deficiency. Mol. Genet. Metab. 2007, 92, 229–233. [Google Scholar] [CrossRef]
- Pingitore, P.; Lepore, S.M.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Valenti, L.; Berge, K.E.; Retterstøl, K.; Leren, T.P.; Wiklund, O.; et al. Identification and Characterization of Two Novel Mutations in the LPL Gene Causing Type I Hyperlipoproteinemia. J. Clin. Lipidol. 2016, 10, 816–823. [Google Scholar] [CrossRef]
- Li, X.; Yang, Q.; Shi, X.; Chen, W.; Pu, N.; Li, W.; Li, J. Compound but Non-Linked Heterozygous p.W14X and p.L279 V LPL Gene Mutations in a Chinese Patient with Long-Term Severe Hypertriglyceridemia and Recurrent Acute Pancreatitis. Lipids Health Dis. 2018, 17, 144. [Google Scholar] [CrossRef]
- Nakamura, T.; Suehiro, T.; Yasuoka, N.; Yamamoto, M.; Ito, H.; Yamano, T.; Hashimoto, K. A Novel Nonsense Mutation in Exon 1 and a Transition in Intron 3 of the Lipoprotein Lipase Gene. J. Atheroscler. Thromb. 1996, 3, 17–24. [Google Scholar] [CrossRef]
- Gin, P.; Goulbourne, C.N.; Adeyo, O.; Beigneux, A.P.; Davies, B.S.J.; Tat, S.; Voss, C.V.; Bensadoun, A.; Fong, L.G.; Young, S.G. Chylomicronemia Mutations Yield New Insights into Interactions between Lipoprotein Lipase and GPIHBP1. Human. Mol. Genet. 2012, 21, 2961–2972. [Google Scholar] [CrossRef]
- Pruneta-Deloche, V.; Marçais, C.; Perrot, L.; Sassolas, A.; Delay, M.; Estour, B.; Lagarde, M.; Moulin, P. Combination of Circulating Antilipoprotein Lipase (Anti-LPL) Antibody and Heterozygous S172 fsX179 Mutation of LPL Gene Leading to Chronic Hyperchylomicronemia. J. Clin. Endocrinol. Metab. 2005, 90, 3995–3998. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Zhao, T.-Q.; Wang, L.; Liu, Z.-P.; Zhang, C.-M.; Chen, R.; Li, L.; Liu, G.; Hu, W.-C. A Novel Substitution at the Translation Initiator Codon (ATG-->ATC) of the Lipoprotein Lipase Gene Is Mainly Responsible for Lipoprotein Lipase Deficiency in a Patient with Severe Hypertriglyceridemia and Recurrent Pancreatitis. Biochem. Biophys. Res. Commun. 2006, 341, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, Y.; Shi, Y.; Zhao, W.; Gao, L.; Fang, L.; Jin, X.; Han, X.; Sun, Q.; Li, G.; et al. Identification and Characterization of Two Novel Compounds: Heterozygous Variants of Lipoprotein Lipase in Two Pedigrees with Type I Hyperlipoproteinemia. Front. Endocrinol. 2022, 13, 874608. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Arce, A.J.; Noderer, W.L.; Wang, C.L. Complete Motif Analysis of Sequence Requirements for Translation Initiation at Non-AUG Start Codons. Nucleic Acids Res. 2018, 46, 985–994. [Google Scholar] [CrossRef]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of Translation Initiation from All 64 Codons in E. Coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef]
- Tada, H.; Nomura, A.; Okada, H.; Nakahashi, T.; Nozue, T.; Hayashi, K.; Nohara, A.; Yagi, K.; Inazu, A.; Michishita, I.; et al. Clinical Whole Exome Sequencing in Severe Hypertriglyceridemia. Clin. Chim. Acta 2019, 488, 31–39. [Google Scholar] [CrossRef]
- Owji, H.; Nezafat, N.; Negahdaripour, M.; Hajiebrahimi, A.; Ghasemi, Y. A Comprehensive Review of Signal Peptides: Structure, Roles, and Applications. Eur. J. Cell Biol. 2018, 97, 422–441. [Google Scholar] [CrossRef]
- Duffy, J.; Patham, B.; Mensa-Wilmot, K. Discovery of Functional Motifs in H-Regions of Trypanosome Signal Sequences. Biochem. J. 2010, 426, 135–145. [Google Scholar] [CrossRef]
- Kobayashi, J.; Inadera, H.; Fujita, Y.; Talley, G.; Morisaki, N.; Yoshida, S.; Saito, Y.; Fojo, S.S.; Brewer, H.B. A Naturally Occurring Mutation at the Second Base of Codon Asparagine 43 in the Proposed N-Linked Glycosylation Site of Human Lipoprotein Lipase: In Vivo Evidence That Asparagine 43 Is Essential for Catalysis and Secretion. Biochem. Biophys. Res. Commun. 1994, 205, 506–515. [Google Scholar] [CrossRef]
- Rabacchi, C.; Pisciotta, L.; Cefalù, A.B.; Noto, D.; Fresa, R.; Tarugi, P.; Averna, M.; Bertolini, S.; Calandra, S. Spectrum of Mutations of the LPL Gene Identified in Italy in Patients with Severe Hypertriglyceridemia. Atherosclerosis 2015, 241, 79–86. [Google Scholar] [CrossRef]
- Kolářová, H.; Tesařová, M.; Švecová, Š.; Stránecký, V.; Přistoupilová, A.; Zima, T.; Uhrová, J.; Volgina, S.Y.; Zeman, J.; Honzík, T. Lipoprotein Lipase Deficiency: Clinical, Biochemical and Molecular Characteristics in Three Patients with Novel Mutations in the LPL Gene. Fol. Biol. 2014, 60, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Roshan, R.; Desai, R.; Kadam, S. Neonatal Hyperlipidemia with Pancreatitis: Novel Gene Mutation of Lipoprotein Lipase. J. Postgrad. Med. 2018, 64, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Bruin, T.; Tuzgöl, S.; Van Diermen, D.E.; Hoogerbrugge-van Der Linden, N.; Brunzell, J.D.; Hayden, M.R.; Kastelein, J.J.P. Recurrent Pancreatitis and Chylomicronemia in an Extended Dutch Kindred Is Caused by a Gly154–>Ser Substitution in Lipoprotein Lipase. J. Lipid Res. 1993, 34, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, E.; Ghayour-Mobarhan, M.; Ferns, G.A.; Pasdar, A. A Novel Variant in LPL Gene Is Associated with Familial Combined Hyperlipidemia. BioFactors 2020, 46, 94–99. [Google Scholar] [CrossRef]
- Ma, Y.H.; Bruin, T.; Tuzgol, S.; Wilson, B.I.; Roederer, G.; Liu, M.S.; Davignon, J.; Kastelein, J.J.; Brunzell, J.D.; Hayden, M.R. Two Naturally Occurring Mutations at the First and Second Bases of Codon Aspartic Acid 156 in the Proposed Catalytic Triad of Human Lipoprotein Lipase. In Vivo Evidence That Aspartic Acid 156 Is Essential for Catalysis. J. Biol. Chem. 1992, 267, 1918–1923. [Google Scholar] [CrossRef]
- Faustinella, F.; Chang, A.; Van Biervliet, J.P.; Rosseneu, M.; Vinaimont, N.; Smith, L.C.; Chen, S.H.; Chan, L. Catalytic Triad Residue Mutation (Asp156—-Gly) Causing Familial Lipoprotein Lipase Deficiency. Co-Inheritance with a Nonsense Mutation (Ser447—-Ter) in a Turkish Family. J. Biol. Chem. 1991, 266, 14418–14424. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, M.S.; Ginzinger, D.; Frohlich, J.; Brunzell, J.D.; Hayden, M.R. Gene-Environment Interaction in the Conversion of a Mild-to-Severe Phenotype in a Patient Homozygous for a Ser172-->Cys Mutation in the Lipoprotein Lipase Gene. J. Clin. Investig. 1993, 91, 1953–1958. [Google Scholar] [CrossRef]
- Guo, D.; Zheng, Y.; Gan, Z.; Guo, Y.; Jiang, S.; Yang, F.; Xiong, F.; Zheng, H. A Heterozygous LMF1 Gene Mutation (c.1523C>T), Combined with an LPL Gene Mutation (c.590G>A), Aggravates the Clinical Symptoms in Hypertriglyceridemia. Front. Genet. 2022, 13, 814295. [Google Scholar] [CrossRef]
- Glueck, C.J.; Levy, R.I.; Glueck, H.I.; Gralnick, H.R.; Greten, H.; Fredrickson, D.S. Acquired Type I Hyperlipoproteinemia with Systemic Lupus Erythematosus, Dysglobulinemia and Heparin Resistance. Am. J. Med. 1969, 47, 318–324. [Google Scholar] [CrossRef]
- Pauciullo, P.; De Simone, B.; Rubba, P.; Mancini, M. A Case of Association between Type I Hyperlipoproteinemia and Systemic Lupus Erythematosus (SLE). Effects of Steroid Treatment. J. Endocrinol. Investig. 1986, 9, 517–520. [Google Scholar] [CrossRef]
- Reichlin, M.; Fesmire, J.; Quintero-Del-Rio, A.I.; Wolfson-Reichlin, M. Autoantibodies to lipoprotein lipase and dyslipidemia in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, J.F.; Borba, E.F.; Viana, V.S.T.; Bueno, C.; Leon, E.P.; Bonfá, E. Anti–Lipoprotein Lipase Antibodies: A New Player in the Complex Atherosclerotic Process in Systemic Lupus Erythematosus? Arthritis Rheum. 2004, 50, 3610–3615. [Google Scholar] [CrossRef]
- Emi, M.; Wilson, D.E.; Iverius, P.H.; Wu, L.; Hata, A.; Hegele, R.; Williams, R.R.; Lalouel, J.M. Missense Mutation (Gly—Glu188) of Human Lipoprotein Lipase Imparting Functional Deficiency. J. Biol. Chem. 1990, 265, 5910–5916. [Google Scholar] [CrossRef] [PubMed]
- Henderson, H.E.; Hassan, F.; Berger, G.M.; Hayden, M.R. The Lipoprotein Lipase Gly188—Glu Mutation in South Africans of Indian Descent: Evidence Suggesting Common Origins and an Increased Frequency. J. Med. Genet. 1992, 29, 119–122. [Google Scholar] [CrossRef]
- Wood, S.; Schertzer, M.; Hayden, M.; Ma, Y. Support for Founder Effect for Two Lipoprotein Lipase (LPL) Gene Mutations in French Canadians by Analysis of GT Microsatellites Flanking the LPL Gene. Hum. Genet. 1993, 91, 312–316. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Y.; He, X.; Deng, C.; Liu, X.; Li, J.; Zhou, L.; Li, Y.; Zhang, Y.; Liu, H.; et al. Two Novel Mutations of the LPL Gene in Two Chinese Family Cases with Familial Chylomicronemia Syndrome. Clin. Chim. Acta 2021, 521, 264–271. [Google Scholar] [CrossRef]
- Chokshi, N.; Blumenschein, S.D.; Ahmad, Z.; Garg, A. Genotype-Phenotype Relationships in Patients with Type I Hyperlipoproteinemia. J. Clin. Lipidol. 2014, 8, 287–295. [Google Scholar] [CrossRef]
- Teramoto, R.; Tada, H.; Kawashiri, M.; Nohara, A.; Nakahashi, T.; Konno, T.; Inazu, A.; Mabuchi, H.; Yamagishi, M.; Hayashi, K. Molecular and Functional Characterization of Familial Chylomicronemia Syndrome. Atherosclerosis 2018, 269, 272–278. [Google Scholar] [CrossRef]
- Kobayashi, J.; Sasaki, N.; Tashiro, J.; Inadera, H.; Saito, Y.; Yoshida, S. A Missense Mutation (Ala334⇒Thr) in Exon 7 of the Lipoprotein Lipase Gene in a Case with Type I Hyperlipidemia. Biochem. Biophys. Res. Commun. 1993, 191, 1046–1054. [Google Scholar] [CrossRef]
- Chan, L.Y.S.; Lam, C.-W.; Mak, Y.-T.; Tomlinson, B.; Tsang, M.-W.; Baum, L.; Masarei, J.R.L.; Pang, C.-P. Genotype-Phenotype Studies of Six Novel LPL Mutations in Chinese Patients with Hypertriglyceridemia. Human. Mutat. 2002, 20, 232–233. [Google Scholar] [CrossRef]
- Lun, Y.; Sun, X.; Wang, P.; Chi, J.; Hou, X.; Wang, Y. Severe Hypertriglyceridemia Due to Two Novel Loss-of-Function Lipoprotein Lipase Gene Mutations (C310R/E396V) in a Chinese Family Associated with Recurrent Acute Pancreatitis. Oncotarget 2017, 8, 47741–47754. [Google Scholar] [CrossRef] [PubMed]
- Wiebusch, H.; Funke, H.; Bruin, T.; Bucher, H.; von Eckardstein, A.; Kastelein, J.J.P.; Assmann, G. Compound Heterozygosity for a Known (D250N) and a Novel (E410K) Missense Mutation in the C-terminal Domain of Lipoprotein Lipase Causes Familial Chylomicronemia. Human. Mutat. 1996, 8, 381–383. [Google Scholar] [CrossRef]
- Previato, L.; Guardamagna, O.; Dugi, K.A.; Ronan, R.; Talley, G.D.; Santamarina-Fojo, S.; Brewer, H.B. A Novel Missense Mutation in the C-terminal Domain of Lipoprotein Lipase (Glu410–>Val) Leads to Enzyme Inactivation and Familial Chylomicronemia. J. Lipid Res. 1994, 35, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.V.; Davies, B.S.J.; Tat, S.; Gin, P.; Fong, L.G.; Pelletier, C.; Mottler, C.D.; Bensadoun, A.; Beigneux, A.P.; Young, S.G. Mutations in Lipoprotein Lipase That Block Binding to the Endothelial Cell Transporter GPIHBP1. Proc. Natl. Acad. Sci. USA 2011, 108, 7980–7984. [Google Scholar] [CrossRef] [PubMed]
- Henderson, H.E.; Hassan, F.; Marais, D.; Hayden, M.R. A New Mutation Destroying Disulphide Bridging in the C-terminal Domain of Lipoprotein Lipase. Biochem. Biophys. Res. Commun. 1996, 227, 189–194. [Google Scholar] [CrossRef]
- Henderson, H.; Leisegang, F.; Hassan, F.; Hayden, M.; Marais, D. A Novel Glu421Lys Substitution in the Lipoprotein Lipase Gene in Pregnancy-Induced Hypertriglyceridemic Pancreatitis. Clin. Chim. Acta 1998, 269, 1–12. [Google Scholar] [CrossRef]
- Yang, W.S.; Nevin, D.N.; Peng, R.; Brunzell, J.D.; Deeb, S.S. A Mutation in the Promoter of the Lipoprotein Lipase (LPL) Gene in a Patient with Familial Combined Hyperlipidemia and Low LPL Activity. Proc. Natl. Acad. Sci. USA 1995, 92, 4462–4466. [Google Scholar] [CrossRef]
- Zhang, Q.; Cavallero, E.; Hoffmann, M.M.; Cavanna, J.; Kay, A.; Charles, A.; Braschi, S.; Marz, W.; Perlemuter, L.; Jacotot, B.; et al. Mutations at the Lipoprotein Lipase Gene Locus in Subjects with Diabetes Mellitus, Obesity and Lipaemia. Clin. Sci. 1997, 93, 335–341. [Google Scholar] [CrossRef]
- Yang, L.-X.; Razzaghi, H.; Hokanson, J.E.; Kamboh, M.I. Identification and Characterization of a Novel 5 Bp Deletion in a Putative Insulin Response Element in the Lipoprotein Lipase Gene. Biochim. Biophys. Acta 2009, 1791, 1057–1065. [Google Scholar] [CrossRef]
- Reymer, P.W.A.; Gagné, E.; Groenemeyer, B.E.; Zhang, H.; Forsyth, I.; Jansen, H.; Seidell, J.C.; Kromhout, D.; Lie, K.E.; Kastelein, J.; et al. A Lipoprotein Lipase Mutation (Asn291Ser) Is Associated with Reduced HDL Cholesterol Levels in Premature Atherosclerosis. Nat. Genet. 1995, 10, 28–34. [Google Scholar] [CrossRef]
- Buscà, R.; Peinado, J.; Vilella, E.; Auwerx, J.; Deeb, S.S.; Vilaró, S.; Reina, M. The Mutant Asn291 → Ser Human Lipoprotein Lipase Is Associated with Reduced Catalytic Activity and Does Not Influence Binding to Heparin. FEBS Lett. 1995, 367, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Syvänne, M.; Antikainen, M.; Ehnholm, S.; Tenkanen, H.; Lahdenperä, S.; Ehnholm, C.; Taskinen, M.R. Heterozygosity for Asn291–>Ser Mutation in the Lipoprotein Lipase Gene in Two Finnish Pedigrees: Effect of Hyperinsulinemia on the Expression of Hypertriglyceridemia. J. Lipid Res. 1996, 37, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, G.S.; Tatt, I.; Salanti, G.; Butterworth, A.S.; Sarwar, N.; van Maarle, M.; Jukema, J.W.; Wiman, B.; Kastelein, J.J.P.; Bennet, A.M.; et al. Seven Lipoprotein Lipase Gene Polymorphisms, Lipid Fractions, and Coronary Disease: A HuGE Association Review and Meta-Analysis. Am. J. Epidemiol. 2008, 168, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, M.J.V.; Bredie, S.J.H.; Boomsma, D.I.; Reymer, P.W.A.; Kastelein, J.J.P.; de Knijff, P.; Demacker, P.N.M.; Stalenhoef, A.F.H.; Havekes, L.M.; Frants, R.R. The Lipoprotein Lipase (Asn291 → Ser) Mutation Is Associated with Elevated Lipid Levels in Families with Familial Combined Hyperlipidaemia. Atherosclerosis 1996, 119, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Yang, W.S.; Nevin, D.N.; Iwasaki, L.; Peng, R.; Brown, B.G.; Brunzell, J.D.; Deeb, S.S. Regulatory Mutations in the Human Lipoprotein Lipase Gene in Patients with Familial Combined Hyperlipidemia and Coronary Artery Disease. J. Lipid Res. 1996, 37, 2627–2637. [Google Scholar] [CrossRef]
- Schmella, M.J.; Ferrell, R.E.; Gallaher, M.J.; Lykins, D.L.; Althouse, A.D.; Roberts, J.M.; Hubel, C.A. The -93T/G LPL Promoter Polymorphism Is Associated With Lower Third-Trimester Triglycerides in Pregnant African American Women. Biol. Res. Nurs. 2015, 17, 429–437. [Google Scholar] [CrossRef]
- Chen, Q.; Razzaghi, H.; Demirci, F.Y.; Kamboh, M.I. Functional Significance of Lipoprotein Lipase HindIII Polymorphism Associated with the Risk of Coronary Artery Disease. Atherosclerosis 2008, 200, 102–108. [Google Scholar] [CrossRef]
- Smith, A.J.P.; Palmen, J.; Putt, W.; Talmud, P.J.; Humphries, S.E.; Drenos, F. Application of Statistical and Functional Methodologies for the Investigation of Genetic Determinants of Coronary Heart Disease Biomarkers: Lipoprotein Lipase Genotype and Plasma Triglycerides as an Exemplar. Human. Mol. Genet. 2010, 19, 3936–3947. [Google Scholar] [CrossRef]
- Rodrigues, R.; Artieda, M.; Tejedor, D.; Martínez, A.; Konstantinova, P.; Petry, H.; Meyer, C.; Corzo, D.; Sundgreen, C.; Klor, H.U.; et al. Pathogenic Classification of LPL Gene Variants Reported to Be Associated with LPL Deficiency. J. Clin. Lipidol. 2016, 10, 394–409. [Google Scholar] [CrossRef]
- Guo, S.; Yang, Z.; Guo, H.; Zhang, J.; Tang, J.; Rui, D.; Ma, R. Association of lipoprotein lipase gene Hind III and S447X polymorphisms in metabolic syndrome patients among Kazakh and Han ethnics from Xinjiang. Zhonghua Liu Xing Bing. Xue Za Zhi 2010, 31, 992–996. [Google Scholar] [PubMed]
- Hata, A.; Robertson, M.; Emi, M.; Lalouel, J.M. Direct Detection and Automated Sequencing of Individual Alleles after Electrophoretic Strand Separation: Identification of a Common Nonsense Mutation in Exon 9 of the Human Lipoprotein Lipase Gene. Nucleic Acids Res. 1990, 18, 5407–5411. [Google Scholar] [CrossRef] [PubMed]
- Elbein, S.C.; Yeager, C.; Kwong, L.K.; Lingam, A.; Inoue, I.; Lalouel, J.M.; Wilson, D.E. Molecular Screening of the Lipoprotein Lipase Gene in Hypertriglyceridemic Members of Familial Noninsulin-Dependent Diabetes Mellitus Families. J. Clin. Endocrinol. Metab. 1994, 79, 1450–1456. [Google Scholar] [CrossRef]
- Matern, D.; Seydewitz, H.; Niederhoff, H.; Wiebusch, H.; Brandis, M. Dyslipidaemia in a Boy with Recurrent Abdominal Pain, Hypersalivation and Decreased Lipoprotein Lipase Activity. Eur. J. Pediatr. 1996, 155, 660–664. [Google Scholar] [CrossRef]
- Gagné, S.; Larson, M.; Pimstone, S.; Schaefer, E.; Kastelein, J.; Wilson, P.; Ordovas, J.; Hayden, M. A Common Truncation Variant of Lipoprotein Lipase (Ser447X) Confers Protection against Coronary Heart Disease: The Framingham Offspring Study. Clin. Genet. 1999, 55, 450–454. [Google Scholar] [CrossRef]
- Karagianni, C.; Stabouli, S.; Roumeliotou, K.; Traeger-Synodinos, J.; Kavazarakis, E.; Gourgiotis, D.; Lambrou, J.; Kanavakis, E. Severe Hypertriglyceridaemia in Diabetic Ketoacidosis: Clinical and Genetic Study. Diabet. Med. 2004, 21, 380–382. [Google Scholar] [CrossRef]
- Liu, A.; Li, L.; Cao, W.; Shan, S.; Lu, J.; Guo, X.; Hu, Y. The association of S447X and Hind III polymorphism in the lipoprotein lipase gene with dyslipidemia of the metabolic syndrome in patients with essential hypertension. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2005, 22, 151–157. [Google Scholar]
- Guan, G.; Xu, E.; Wang, X.; Xu, Y.; Qiu, S. Associations between Ser447Ter Gene Polymorphism of Lipoprotein Lipase and Atherosclerotic Cerebral Infarction. Zhonghua Yi Xue Yi Chuan Xue Za Zhi = Zhonghua Yixue Yichuanxue Zazhi = Chin. J. Med. Genet. 2006, 23, 519–522. [Google Scholar]
- Herbeth, B.; Gueguen, S.; Leroy, P.; Siest, G.; Visvikis-Siest, S. The Lipoprotein Lipase Serine 447 Stop Polymorphism Is Associated with Altered Serum Carotenoid Concentrations in the Stanislas Family Study. J. Am. Coll. Nutr. 2007, 26, 655–662. [Google Scholar] [CrossRef]
- Komurcu-Bayrak, E.; Onat, A.; Poda, M.; Humphries, S.E.; Acharya, J.; Hergenc, G.; Coban, N.; Can, G.; Erginel-Unaltuna, N. The S447X Variant of Lipoprotein Lipase Gene Is Associated with Metabolic Syndrome and Lipid Levels among Turks. Clin. Chim. Acta 2007, 383, 110–115. [Google Scholar] [CrossRef]
- Talmud, P.J.; Flavell, D.M.; Alfakih, K.; Cooper, J.A.; Balmforth, A.J.; Sivananthan, M.; Montgomery, H.E.; Hall, A.S.; Humphries, S.E. The Lipoprotein Lipase Gene Serine 447 Stop Variant Influences Hypertension-Induced Left Ventricular Hypertrophy and Risk of Coronary Heart Disease. Clin. Sci. 2007, 112, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mu, Y.; Zhao, Y.; Liu, X.; Zhao, L.; Wang, J.; Xie, Y. Genetic Screening of the Lipoprotein Lipase Gene for Mutations in Chinese Subjects with or without Hypertriglyceridemia. J. Genet. Genom. 2007, 34, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Aydogan, H.Y.; Isbïr, S.; Kurnaz, O.; Gormus, U.; Isbïr, T. Associations of Lipoprotein Lipase S447X and Apolipoprotein E Genotypes with Low-Density Lipoprotein Subfractions in Turkish Patients with Coronary Artery Disease. In Vivo 2009, 23, 155–161. [Google Scholar] [PubMed]
- Chang, Y.-T.; Chang, M.-C.; Su, T.-C.; Liang, P.-C.; Su, Y.-N.; Kuo, C.-H.; Wei, S.-C.; Wong, J.-M. Lipoprotein Lipase Mutation S447X Associated with Pancreatic Calcification and Steatorrhea in Hyperlipidemic Pancreatitis. J. Clin. Gastroenterol. 2009, 43, 591–596. [Google Scholar] [CrossRef]
- Deo, R.C.; Reich, D.; Tandon, A.; Akylbekova, E.; Patterson, N.; Waliszewska, A.; Kathiresan, S.; Sarpong, D.; Taylor, H.A.; Wilson, J.G. Genetic Differences between the Determinants of Lipid Profile Phenotypes in African and European Americans: The Jackson Heart Study. PLoS Genet. 2009, 5, e1000342. [Google Scholar] [CrossRef]
- Fujiwara, S.; Kotani, K.; Sano, Y.; Matsuoka, Y.; Tsuzaki, K.; Domichi, M.; Kajii, E.; Sakane, N. S447X Polymorphism in the Lipoprotein Lipase Gene and the Adiponectin Level in the General Population: Results from the Mima Study. JAT 2009, 16, 188–193. [Google Scholar] [CrossRef]
- Jensen, M.K.; Rimm, E.B.; Rader, D.; Schmidt, E.B.; Sørensen, T.I.A.; Vogel, U.; Overvad, K.; Mukamal, K.J. S447X Variant of the Lipoprotein Lipase Gene, Lipids, and Risk of Coronary Heart Disease in 3 Prospective Cohort Studies. Am. Heart J. 2009, 157, 384–390. [Google Scholar] [CrossRef]
- Salah, A.; Khan, M.; Esmail, N.; Habibullah, S.; Al Lahham, Y. Genetic Polymorphism of S447X Lipoprotein Lipase (LPL) and the Susceptibility to Hypertension. J. Crit. Care 2009, 24, e11–e14. [Google Scholar] [CrossRef]
- van Hoek, M.; Dallinga-Thie, G.M.; Steyerberg, E.W.; Sijbrands, E.J.G. Diagnostic Value of Post-Heparin Lipase Testing in Detecting Common Genetic Variants in the LPL and LIPC Genes. Eur. J. Hum. Genet. 2009, 17, 1386–1393. [Google Scholar] [CrossRef]
- Webster, R.J.; Warrington, N.M.; Weedon, M.N.; Hattersley, A.T.; McCaskie, P.A.; Beilby, J.P.; Palmer, L.J.; Frayling, T.M. The Association of Common Genetic Variants in the APOA5, LPL and GCK Genes with Longitudinal Changes in Metabolic and Cardiovascular Traits. Diabetologia 2009, 52, 106–114. [Google Scholar] [CrossRef]
- Wang, C.; Sun, T.; Li, H.; Bai, J.; Li, Y. Lipoprotein Lipase Ser447Ter Polymorphism Associated with the Risk of Ischemic Stroke: A Meta-Analysis. Thromb. Res. 2011, 128, e107–e112. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, G.; Unal, R.; Pokrovskaya, I.D.; Tripathi, P.; Rotter, J.I.; Goodarzi, M.O.; Kern, P.A. The Lipoprotein Lipase (LPL) S447X Gain of Function Variant Involves Increased mRNA Translation. Atherosclerosis 2011, 221, 143. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; Isono, M.; Katsuya, T.; Yokota, M.; Yamamoto, K.; Nabika, T.; Shimokawa, K.; Nakashima, E.; Sugiyama, T.; Rakugi, H.; et al. Association of Genetic Variants Influencing Lipid Levels with Coronary Artery Disease in Japanese Individuals. PLoS ONE 2012, 7, e46385. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Towne, M.C.; Luquette, L.J.; Harris, D.J.; Marinakis, N.S.; Meinecke, P.; Kutsche, K.; Campeau, P.M.; Yu, T.W.; Margulies, D.M.; et al. Mutation of KCNJ8 in a Patient with Cantú Syndrome with Unique Vascular Abnormalities—Support for the Role of K(ATP) Channels in This Condition. Eur. J. Med. Genet. 2013, 56, 678–682. [Google Scholar] [CrossRef]
- Velapasamy, S.; Alex, L.; Chahil, J.K.; Lye, S.H.; Munretnam, K.; Nor Hashim, N.A.; Ramzi, N.H.; Mohd Nordin, N.; Visvalingam, V.; Ler, L.W. Influences of Multiple Genetic Polymorphisms on Ovarian Cancer Risk in Malaysia. Genet. Test. Mol. Biomark. 2013, 17, 62–68. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Wang, L.; Liao, Q.; Chang, L.; Xu, L.; Huang, Y.; Ye, H.; Xu, L.; Chen, C.; et al. Meta-Analyses of 8 Polymorphisms Associated with the Risk of the Alzheimer’s Disease. PLoS ONE 2013, 8, e73129. [Google Scholar] [CrossRef]
- Bentley, A.R.; Chen, G.; Shriner, D.; Doumatey, A.P.; Zhou, J.; Huang, H.; Mullikin, J.C.; Blakesley, R.W.; Hansen, N.F.; Bouffard, G.G.; et al. Gene-Based Sequencing Identifies Lipid-Influencing Variants with Ethnicity-Specific Effects in African Americans. PLoS Genet. 2014, 10, e1004190. [Google Scholar] [CrossRef]
- Turlo, K.; Leung, C.S.; Seo, J.J.; Goulbourne, C.N.; Adeyo, O.; Gin, P.; Voss, C.; Bensadoun, A.; Fong, L.G.; Young, S.G.; et al. Equivalent Binding of Wild-Type Lipoprotein Lipase (LPL) and S447X-LPL to GPIHBP1, the Endothelial Cell LPL Transporter. Biochim. Biophys. Acta 2014, 1841, 963. [Google Scholar] [CrossRef]
- Zambrano Morales, M.; Fernández Salgado, E.; Balzán Urdaneta, L.; Labastidas, N.; Aranguren-Méndez, J.; Connell, L.; Molero Paredes, T.; Rojas, A.; Panunzio, A. Lack of association between the S447X variant of the lipoprotein lipase gene and plasma lipids. A preliminary study. Investig. Clin. 2014, 55, 133–141. [Google Scholar]
- Emamian, M.; Avan, A.; Pasdar, A.; Mirhafez, S.R.; Sadeghzadeh, M.; Moghadam, M.S.; Parizadeh, S.M.R.; Ferns, G.A.; Ghayour-Mobarhan, M. The Lipoprotein Lipase S447X and Cholesteryl Ester Transfer Protein Rs5882 Polymorphisms and Their Relationship with Lipid Profile in Human Serum of Obese Individuals. Gene 2015, 558, 195–199. [Google Scholar] [CrossRef]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and Clinical Impact of Functional Variants in 50,726 Whole-Exome Sequences from the DiscovEHR Study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, J.; Li, H.; Chen, Y.; Wang, L.; He, M.; Wang, Y.; Sun, L.; Hu, Y.; Huang, J.; et al. Coding-Sequence Variants Are Associated with Blood Lipid Levels in 14,473 Chinese. Human. Mol. Genet. 2016, 25, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Ren, X. Meta-Analyses of Four Polymorphisms of Lipoprotein Lipase Associated with the Risk of Alzheimer’s Disease. Neurosci. Lett. 2016, 619, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Shatwan, I.M.; Minihane, A.-M.; Williams, C.M.; Lovegrove, J.A.; Jackson, K.G.; Vimaleswaran, K.S. Impact of Lipoprotein Lipase Gene Polymorphism, S447X, on Postprandial Triacylglycerol and Glucose Response to Sequential Meal Ingestion. Int. J. Mol. Sci. 2016, 17, 397. [Google Scholar] [CrossRef]
- Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators; Stitziel, N.O.; Stirrups, K.E.; Masca, N.G.D.; Erdmann, J.; Ferrario, P.G.; König, I.R.; Weeke, P.E.; Webb, T.R.; Auer, P.L.; et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N. Engl. J. Med. 2016, 374, 1134–1144. [Google Scholar] [CrossRef]
- Verma, A.; Verma, S.S.; Pendergrass, S.A.; Crawford, D.C.; Crosslin, D.R.; Kuivaniemi, H.; Bush, W.S.; Bradford, Y.; Kullo, I.; Bielinski, S.J.; et al. eMERGE Phenome-Wide Association Study (PheWAS) Identifies Clinical Associations and Pleiotropy for Stop-Gain Variants. BMC Med. Genom. 2016, 9 (Suppl. S1), 32. [Google Scholar] [CrossRef]
- Hayne, C.K.; Lafferty, M.J.; Eglinger, B.J.; Kane, J.P.; Neher, S.B. Biochemical Analysis of the Lipoprotein Lipase Truncation Variant, LPLS447X, Reveals Increased Lipoprotein Uptake. Biochemistry 2017, 56, 525–533. [Google Scholar] [CrossRef]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-Wide Association Study of Plasma Lipids in >300,000 Individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef]
- Lu, X.; Peloso, G.M.; Liu, D.J.; Wu, Y.; Zhang, H.; Zhou, W.; Li, J.; Tang, C.S.-M.; Dorajoo, R.; Li, H.; et al. Exome Chip Meta-Analysis Identifies Novel Loci and East Asian-Specific Coding Variants That Contribute to Lipid Levels and Coronary Artery Disease. Nat. Genet. 2017, 49, 1722–1730. [Google Scholar] [CrossRef]
- Wang, J.; Du, S.; Wang, J.; Zhu, M.; Wen, X.; Yang, W. Association of the Lipoprotein Lipase Gene Ser447Ter Polymorphism with Hypertension and Blood Pressure Variation: Evidence from an Updated Meta-Analysis. Clin. Exp. Hypertens. 2017, 39, 655–664. [Google Scholar] [CrossRef]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wu, Y.; Wen, Y.; Guo, M.; Zhang, H. The Association of the S447X Mutation in LPL with Coronary Artery Disease: A Meta-Analysis. Minerva Cardioangiol. 2019, 67, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Marmontel, O.; Rollat-Farnier, P.A.; Wozny, A.-S.; Charrière, S.; Vanhoye, X.; Simonet, T.; Chatron, N.; Collin-Chavagnac, D.; Nony, S.; Dumont, S.; et al. Development of a New Expanded Next-Generation Sequencing Panel for Genetic Diseases Involved in Dyslipidemia. Clin. Genet. 2020, 98, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Rom, O.; Surakka, I.; Graham, S.E.; Zhou, W.; Roychowdhury, T.; Fritsche, L.G.; Gagliano Taliun, S.A.; Sidore, C.; Liu, Y.; et al. Loss-of-Function Genomic Variants Highlight Potential Therapeutic Targets for Cardiovascular Disease. Nat. Commun. 2020, 11, 6417. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.S.; Li, X.; Li, Z.; Pampana, A.; Zhang, D.Y.; Park, J.; Aslibekyan, S.; Bis, J.C.; Brody, J.A.; Cade, B.E.; et al. Whole Genome Sequence Analysis of Blood Lipid Levels in >66,000 Individuals. Nat. Commun. 2022, 13, 5995. [Google Scholar] [CrossRef]
- Rip, J.; Nierman, M.C.; Ross, C.J.; Jukema, J.W.; Hayden, M.R.; Kastelein, J.J.P.; Stroes, E.S.G.; Kuivenhoven, J.A. Lipoprotein Lipase S447X: A Naturally Occurring Gain-of-Function Mutation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1236–1245. [Google Scholar] [CrossRef]
- Caussy, C.; Charrière, S.; Meirhaeghe, A.; Dallongeville, J.; Lefai, E.; Rome, S.; Cuerq, C.; Euthine, V.; Delay, M.; Marmontel, O.; et al. Multiple microRNA Regulation of Lipoprotein Lipase Gene Abolished by 3′UTR Polymorphisms in a Triglyceride-Lowering Haplotype Harboring p.Ser474Ter. Atherosclerosis 2016, 246, 280–286. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Wong, H.; Quiñones, M.J.; Taylor, K.D.; Guo, X.; Castellani, L.W.; Antoine, H.J.; Yang, H.; Hsueh, W.A.; Rotter, J.I. The 3′ Untranslated Region of the Lipoprotein Lipase Gene: Haplotype Structure and Association with Post-Heparin Plasma Lipase Activity. J. Clin. Endocrinol. Metab. 2005, 90, 4816–4823. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
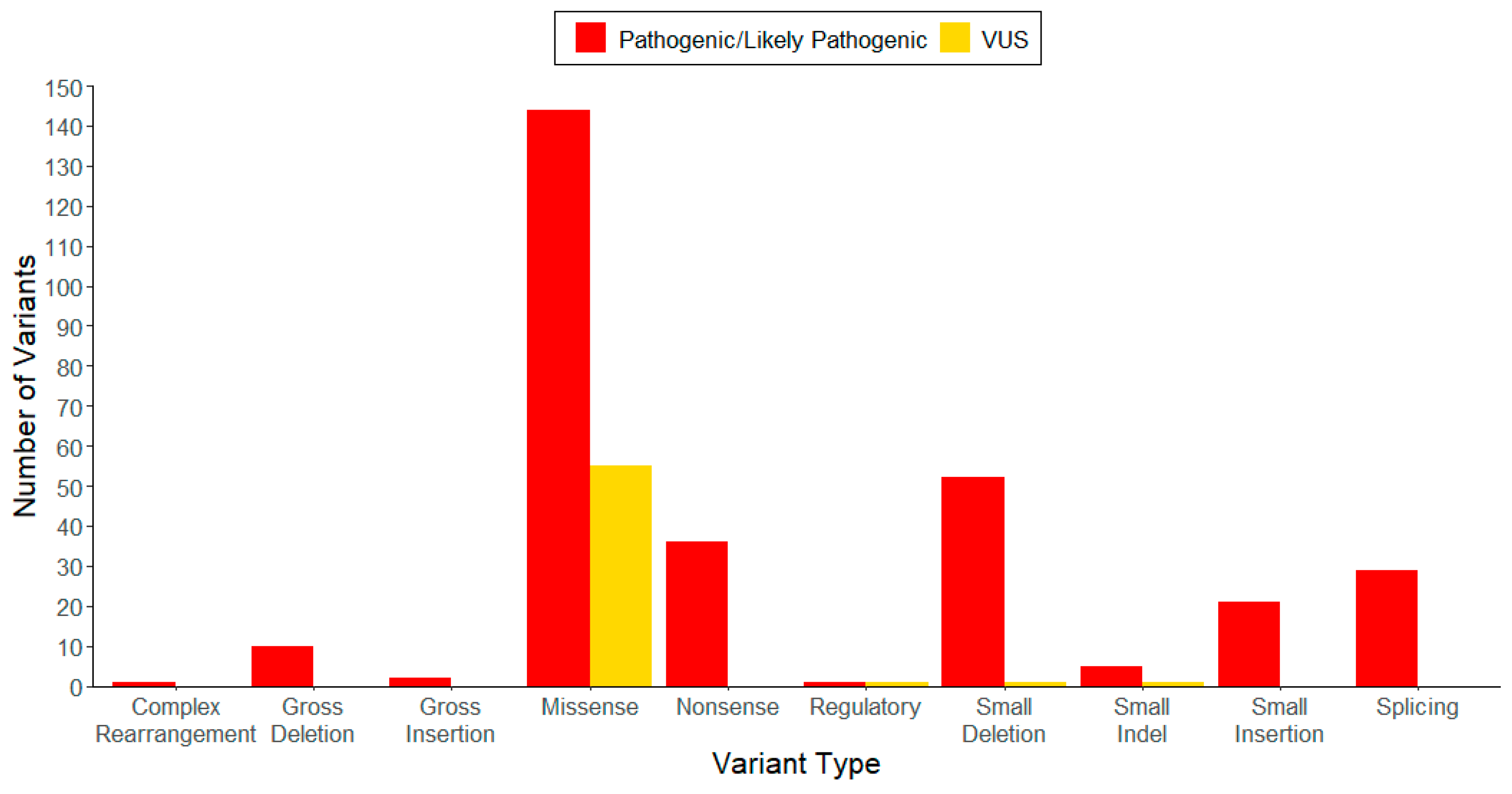
| Variant Type (P-LP 1/VUS 2) | ||||||
|---|---|---|---|---|---|---|
| Genomic Location | Missense | Nonsense | Small Deletion | Small Insertion | Small Indel | Total |
| Exon 1 | 4/1 | 2/0 | 3 */0 | 2/0 | 1/0 | 12/1 |
| Exon 2 | 4/10 | 2/0 | 6/0 | 5/0 | 0/0 | 17/10 |
| Exon 3 | 18/4 | 7/0 | 6/0 | 4/0 | 3/0 | 38/4 |
| Exon 4 | 11/2 | 1/0 | 4/0 | 0/0 | 0/0 | 16/2 |
| Exon 5 | 51/10 | 4/0 | 11/0 | 1/0 | 0/0 | 67/10 |
| Exon 6 | 46/14 | 10/0 | 12/0 | 4/0 | 0/0 | 72/14 |
| Exon 7 | 2/7 | 0/0 | 3/0 | 0/0 | 0/0 | 5/7 |
| Exon 8 | 5/5 | 10/0 | 5/1 | 3/0 | 0/1 | 23/7 |
| Exon 9 | 3/2 | 0/0 | 1/0 | 0/0 | 0/0 | 4/2 |
| Total | 144/55 | 36/0 | 51/1 | 19/0 | 4/1 | 254/57 |
| Variant Type (P-LP 1/VUS 2) | |||||
|---|---|---|---|---|---|
| Genomic Location | Splicing | Small Deletion | Small Insertion | Other | Total |
| Regulatory (Promoter, 5′ UTR, and 3′ UTR) | N/A 3 | 0/0 | 0/0 | 1/1 | 1/1 |
| Intron 1 | 7/0 | 1/0 | 1/0 | N/A | 9/0 |
| Intron 2 | 5 */0 | 0/0 | 1/0 | N/A | 6/0 |
| Intron 3 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Intron 4 | 2/0 | 0/0 | 0/0 | N/A | 2/0 |
| Intron 5 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Intron 6 | 5/0 | 0/0 | 0/0 | N/A | 5/0 |
| Intron 7 | 2/0 | 0/0 | 0/0 | N/A | 2/0 |
| Intron 8 | 5/0 | 0/0 | 0/0 | N/A | 5/0 |
| Intron 9 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Total | 29/0 | 1/0 | 2/0 | 1/0 | 33/1 |
| Variant Type (P-LP 1/VUS 2) | ||
|---|---|---|
| Gross Deletions | Gross Insertions | Complex Rearrangements |
| 10/0 | 2/0 | 1/0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, S.D.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Lipoprotein Lipase: Structure, Function, and Genetic Variation. Genes 2025, 16, 55. https://doi.org/10.3390/genes16010055
Perera SD, Wang J, McIntyre AD, Hegele RA. Lipoprotein Lipase: Structure, Function, and Genetic Variation. Genes. 2025; 16(1):55. https://doi.org/10.3390/genes16010055
Chicago/Turabian StylePerera, Shehan D., Jian Wang, Adam D. McIntyre, and Robert A. Hegele. 2025. "Lipoprotein Lipase: Structure, Function, and Genetic Variation" Genes 16, no. 1: 55. https://doi.org/10.3390/genes16010055
APA StylePerera, S. D., Wang, J., McIntyre, A. D., & Hegele, R. A. (2025). Lipoprotein Lipase: Structure, Function, and Genetic Variation. Genes, 16(1), 55. https://doi.org/10.3390/genes16010055