
Combined Effect of Conventional Chemotherapy with Epigenetic Modulators on Glioblastoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Treatments
2.3. Cytotoxicity Assays and Cell Viability (MTS)
2.4. Cell Cycle Analysis by Flow Cytometry
2.5. Apoptosis Analysis by Flow Cytometry
2.6. Evaluation of Cytokine Levels by ELISA
2.7. RNA Isolation and cDNA Synthesis
2.8. Quantitative Analysis of Selected Genes and lncRNA
2.9. DNA Isolation and LINE-1 Global Methylation Assay
2.10. Statistical Analysis
3. Results
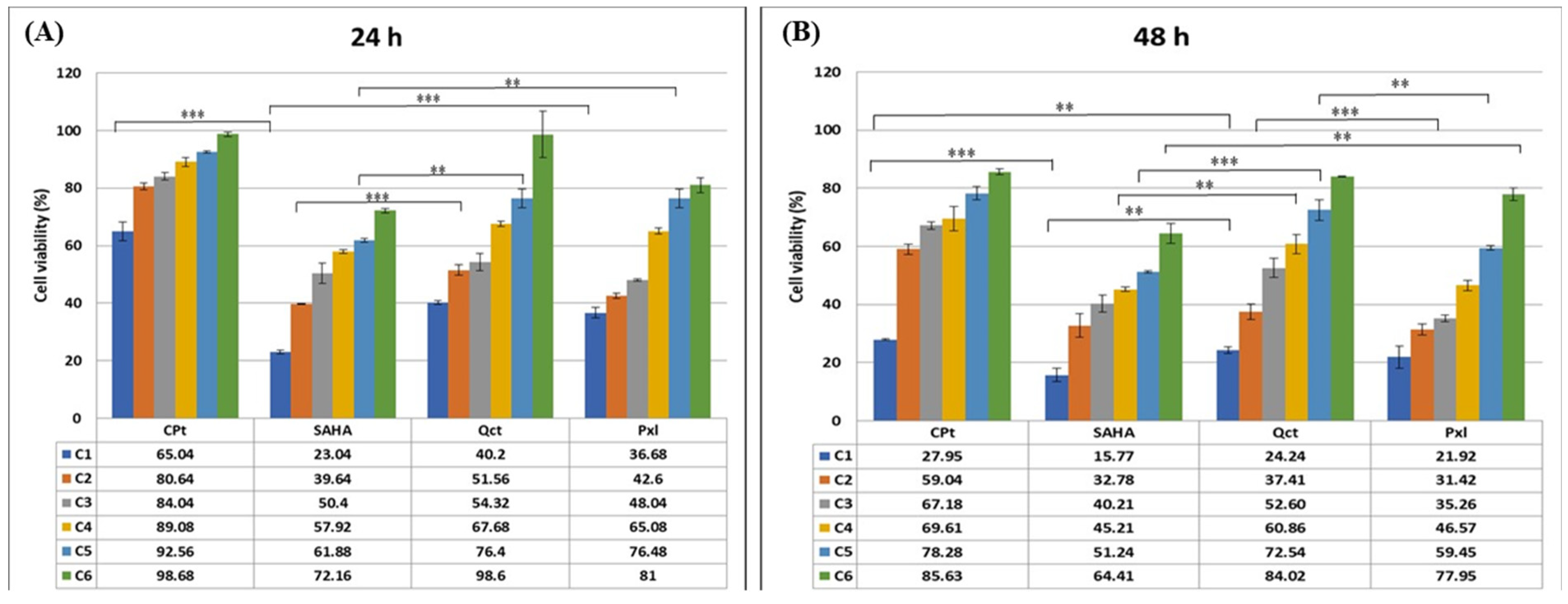
3.1. Drug-Induced Cell Cytotoxicity Is Dose- and Time-Dependent
3.2. Modulation of Cell Cycle Phases in Treated U87-MG Glioma Cells
3.3. Apoptosis Analysis in Treated U87-MG Glioma Cells
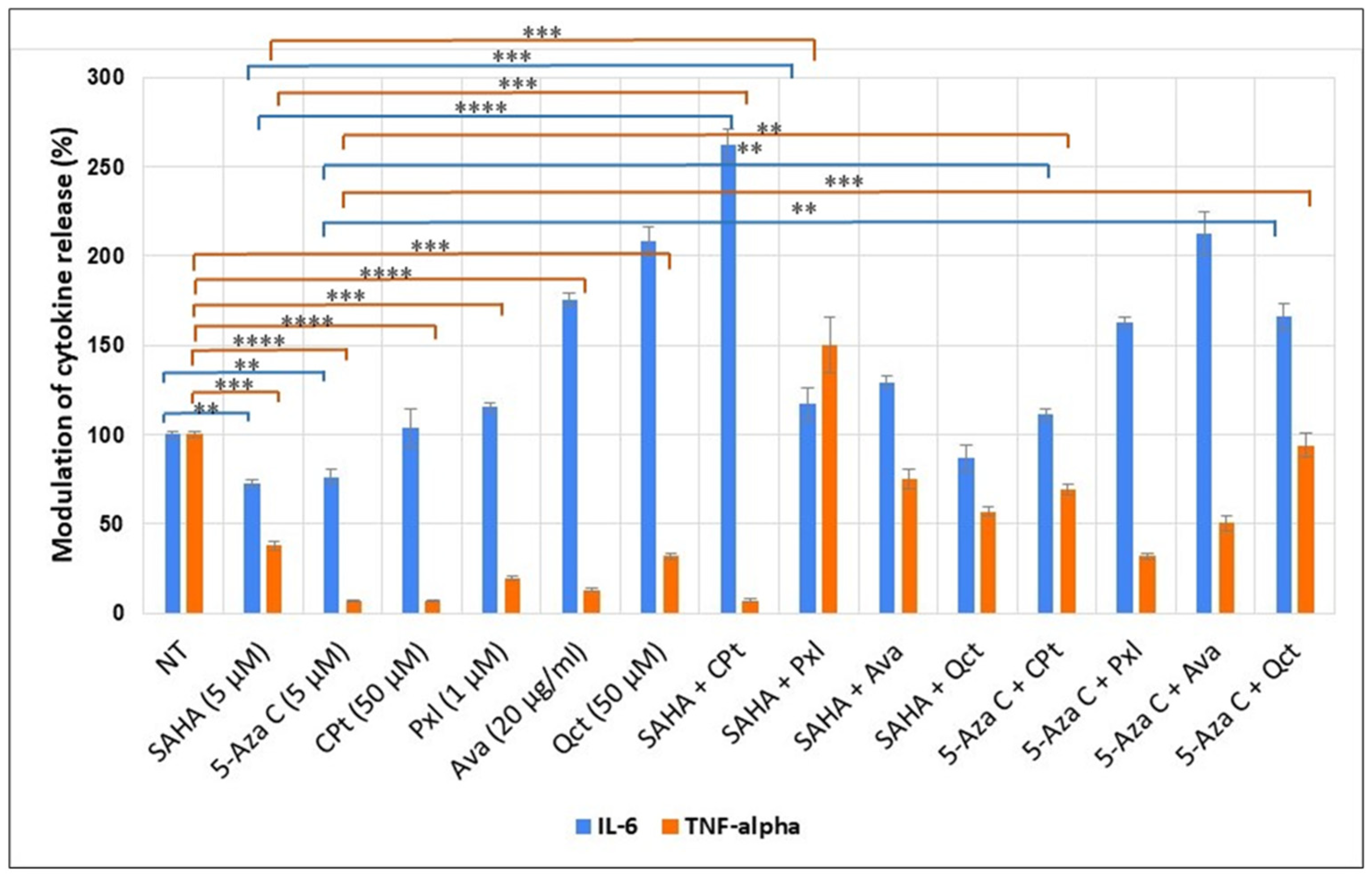
3.4. Evaluation of Soluble Cytokines Released by Treated U87-MG Cells
3.5. Target lncRNAs Gene Expression Quantification
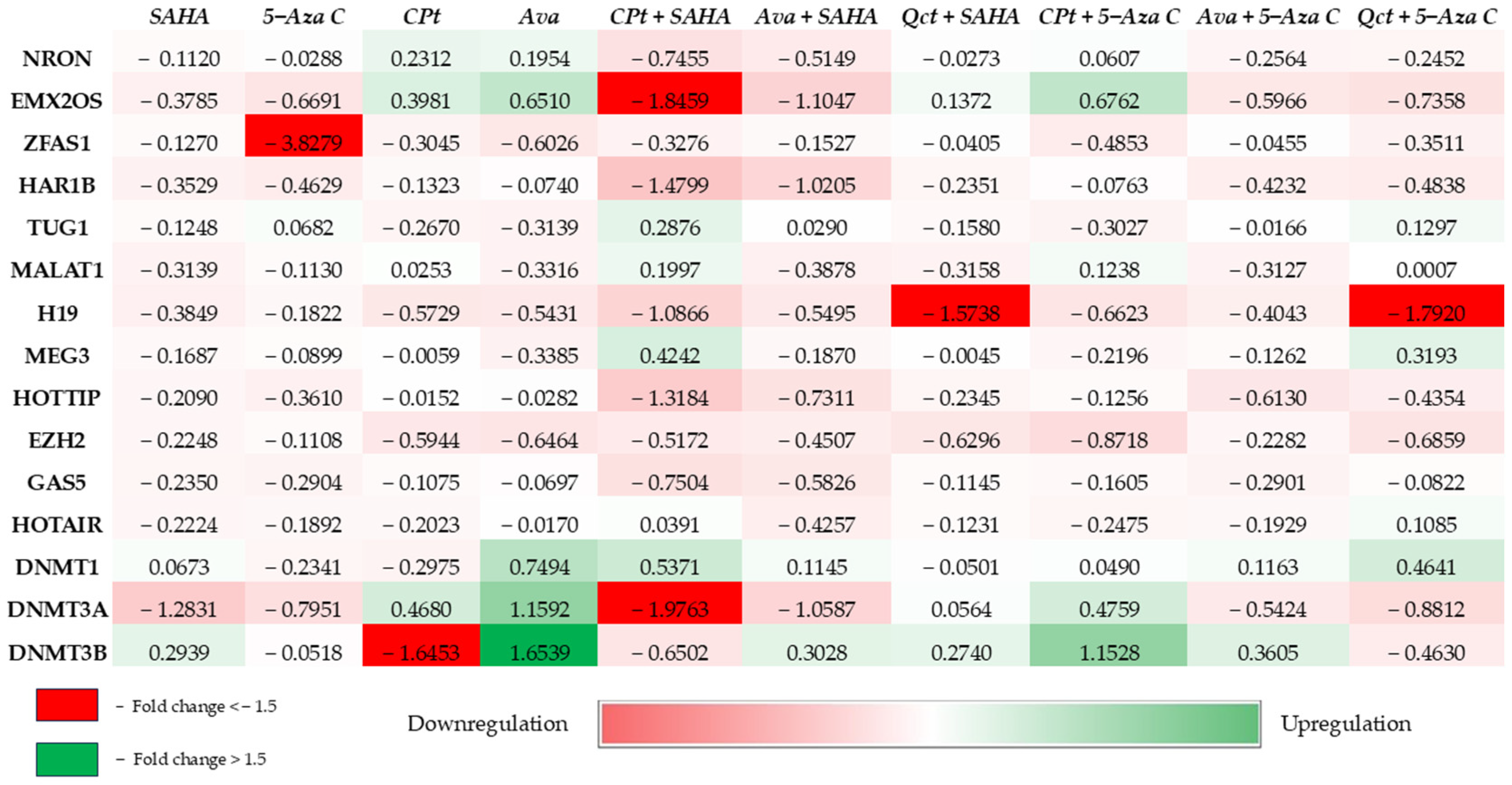
3.6. Epigenetic Modulators Exhibited Modified Expression Pattern and Global Methylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Cho, H.; Mariotto, A.B.; Schwartz, L.M.; Luo, J.; Woloshin, S. When do changes in cancer survival mean progress? The insight from population incidence and mortality. J. Natl. Cancer Inst. Monogr. 2014, 2014, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cancer Trends Progress Report National Cancer Institute; NIH, DHHS, Bethesda, MD. March 2024. Available online: https://progressreport.cancer.gov (accessed on 8 December 2024).
- Krex, D.; Klink, B.; Hartmann, C.; von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. Long-term survival with glioblastoma multiforme. Brain 2007, 130 Pt 10, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P.; (UK) National Cancer Information Network Brain Tumour Group. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Zreik, J.; Moinuddin, F.M.; Yolcu, Y.U.; Alvi, M.A.; Chaichana, K.L.; Quinones-Hinojosa, A.; Bydon, M. Improved 3-year survival rates for glioblastoma multiforme are associated with trends in treatment: Analysis of the national cancer database from 2004 to 2013. J. Neurooncol. 2020, 148, 69–79. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22 (Suppl. S2), iv1–iv96. [Google Scholar] [CrossRef]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma-molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma [Internet]; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, AU, Australia, 2017; Chapter 8. [Google Scholar]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Albulescu, A.; Plesa, A.; Fudulu, A.; Iancu, I.V.; Anton, G.; Botezatu, A. Epigenetic approaches for cervical neoplasia screening (Review). Exp. Ther. Med. 2021, 22, 1481. [Google Scholar] [CrossRef]
- Chen, J.; Ao, L.; Yang, J. Long non-coding RNAs in diseases related to inflammation and immunity. Ann. Transl. Med. 2019, 7, 494. [Google Scholar] [CrossRef]
- Richard Boland, C. Non-coding RNA: It’s Not Junk. Dig. Dis. Sci. 2017, 62, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liu, C.; Wu, M. New insights into long noncoding RNAs and their roles in glioma. Mol. Cancer 2018, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, R.; Sedano, M.J.; Harrison, A.L.; Subramani, R.; Lin, K.Y.; Ramos, E.I.; Lakshmanaswamy, R.; Gadad, S.S. Long noncoding RNAs in cancer: From discovery to therapeutic targets. Adv. Clin. Chem. 2020, 95, 105–147. [Google Scholar] [CrossRef]
- Bjørklund, S.S.; Aure, M.R.; Häkkinen, J.; Vallon-Christersson, J.; Kumar, S.; Evensen, K.B.; Fleischer, T.; Tost, J.; Osbreac; Bathen, T.F.; et al. Subtype and cell type specific expression of lncRNAs provide insight into breast cancer. Commun. Biol. 2022, 5, 834. [Google Scholar] [CrossRef] [PubMed]
- Sousa, G.F.; Wlodarczyk, S.R.; Monteiro, G. Carboplatin: Molecular mechanisms of action associated with chemoresistance. Braz. J. Pharm. 2014, 50, 693–701. [Google Scholar] [CrossRef]
- Jatoi, A.; Martenson, J.A.; Foster, N.R.; McLeod, H.L.; Lair, B.S.; Nichols, F.; Tschetter, L.K.; Moore DFJr Fitch, T.R.; Alberts, S.R.; North Central Cancer Treatment Group (N0044). Paclitaxel, carboplatin, 5-fluorouracil, and radiation for locally advanced esophageal cancer: Phase II results of preliminary pharmacologic and molecular efforts to mitigate toxicity and predict outcomes: North Central Cancer Treatment Group (N0044). Am. J. Clin. Oncol. 2007, 30, 507–513. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Vafadar, A.; Shabaninejad, Z.; Movahedpour, A.; Fallahi, F.; Taghavipour, M.; Ghasemi, Y.; Akbari, M.; Shafiee, A.; Hajighadimi, S.; Moradizarmehri, S.; et al. Quercetin and cancer: New insights into its therapeutic effects on ovarian cancer cells. Cell Biosci. 2020, 10, 32. [Google Scholar] [CrossRef]
- Albulescu, A.; Fudulu, A.; Botezatu, A.; Iancu, I.V.; Plesa, A.; Brasoveanu, L.I. Long noncoding RNA profile in neuroblastoma and glioblastoma. FEBS Open Bio 2021, 12, 83–84. [Google Scholar]
- Albulescu, A.; Petrescu, G.E.D.; Botezatu, A.; Fudulu, A.; Iancu, I.V.; Plesa, A.; Radu, R.; Brehar, F.M.; Gorgan, R.M.; Brasoveanu, L.I. lncRNA Profile in Glioblastoma Samples and Cell Lines. “2nd OncoHUB Conference”. In Proceedings of the 2nd Edition of the OncoHub Conference—Connecting Scientists and Physicians for Next Generation, Brasov, Romania, 21–23 September 2022; Volume 5. Issue 1. [Google Scholar] [CrossRef]
- Mihaila, M.; Bostan, M.; Hotnog, D.; Ferdes, M.; Brasoveanu, L.I. Real-time analysis of quercetin, resveratrol and/or doxorubicin effects in MCF-7 cells. Rom. Biotechnol. Lett. 2013, 18, 8106–8114. [Google Scholar]
- Bostan, M.; Petrica-Matei, G.G.; Ion, G.; Radu, N.; Mihaila, M.; Hainarosie, R.; Brasoveanu, L.I.; Roman, V.; Constantin, C.; Neagu, M.T. Cisplatin effect on head and neck squamous cell carcinoma cells is modulated by ERK1/2 protein kinases. Exp. Ther. Med. 2019, 18, 5041–5051. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, A.-C.; Badea, M.; Olar, R.; Silvestro, L.; Mihaila, M.; Brasoveanu, L.I.; Musat, M.G.; Andries, A.; Uivarosi, V. Cytotoxicity studies, DNA interaction and protein binding of new Al (III), Ga (III) and In (III) complexes with 5-hydroxyflavone. Appl. Organometal Chem. 2018, 32, e4579. [Google Scholar] [CrossRef]
- Munteanu, A.; Musat, M.G.; Mihaila, M.; Badea, M.; Olar, R.; Nitulescu, G.M.; Rădulescu, F.Ș.; Brasoveanu, L.I.; Uivarosi, V. New Heteroleptic Lanthanide Complexes as Multimodal Drugs: Cytotoxicity Studies, Apoptosis, Cell Cycle Analysis, DNA Interactions, and Protein Binding. Appl. Organomet. Chem. 2021, 35, e6062. [Google Scholar] [CrossRef]
- Iancu, I.V.; Botezatu, A.; Plesa, A.; Huica, I.; Fudulu, A.; Albulescu, A.; Bostan, M.; Mihaila, M.; Grancea, C.; Manda, D.A.; et al. Alterations of regulatory factors and DNA methylation pattern in thyroid cancer. Cancer Biomark. 2020, 28, 255–268. [Google Scholar] [CrossRef]
- Ivan, B.-C.; Barbuceanu, S.-F.; Hotnog, C.M.; Anghel, A.I.; Ancuceanu, R.V.; Mihaila, M.A.; Brasoveanu, L.I.; Shova, S.; Draghici, C.; Olaru, O.T.; et al. New Pyrrole Derivatives as Promising Biological Agents: Design, Synthesis, Characterization, In Silico, and Cytotoxicity Evaluation. Int. J. Mol. Sci. 2022, 23, 8854. [Google Scholar] [CrossRef]
- Maciuca, A.-M.; Munteanu, A.-C.; Mihaila, M.; Badea, M.; Olar, R.; Nitulescu, G.M.; Munteanu, C.V.A.; Bostan, M.; Uivarosi, V. Rare-Earth Metal Complexes of the Antibacterial Drug Oxolinic Acid: Synthesis, Characterization, DNA/Protein Binding and Cytotoxicity Studies. Molecules 2020, 25, 5418. [Google Scholar] [CrossRef]
- Fischer, V.; Kalbitz, M.; Müller-Graf, F.; Gebhard, F.; Ignatius, A.; Liedert, A.; Haffner-Luntzer, M. Influence of menopause on inflammatory cytokines during murine and human bone fracture healing. Int. J. Mol. Sci. 2018, 19, 2070. [Google Scholar] [CrossRef]
- Hotnog, D.; Mihaila, M.; Botezatu, A.; Matei, G.G.; Hotnog, C.; Anton, G.; Bostan, M.; Brasoveanu, L.I. Genistein potentiates the apoptotic effect of 5-fluorouracyl in colon cancer cell lines. Rom. Biotechnol. Lett. 2013, 18, 8751–8760. [Google Scholar]
- Feng, Y.; Kang, Y.; He, Y.; Liu, J.; Liang, B.; Yang, P.; Yu, Z. MicroRNA-99a acts as a tumor suppressor and is down-regulated in bladder cancer. BMC Urol. 2014, 14, 50. [Google Scholar] [CrossRef]
- Mohammadi, K.; Baghini, S.S.; Saremi, M.A. Increased expression of the lncH19 gene in the plasma of people with breast cancer. Pers. Med. 2020, 5, 12–14. [Google Scholar] [CrossRef]
- Hotoboc, I.E.; Fudulu, A.; Grigore, R.; Bertesteanu, S.; Huica, I.; Iancu, I.V.; Botezatu, A.; Bleotu, C.; Anton, G. The association between lncRNA H19 and EZH2 expression in patients with EBV-positive laryngeal carcinoma. Acta Otorhinolaryngol. Ital. 2021, 41, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Panoilia, E.; Schindler, E.; Samantas, E.; Aravantinos, G.; Kalofonos, H.P.; Christodoulou, C.; Patrinos, G.P.; Friberg, L.E.; Sivolapenko, G. A pharmacokinetic binding model for bevacizumab and VEGF165 in colorectal cancer patients. Cancer Chemother. Pharmacol. 2015, 75, 791–803. [Google Scholar] [CrossRef]
- Albulescu, A.; Petrescu, G.E.D.; Fudulu, A.; Botezatu, A.; Iancu, I.V.; Pleșa, A.; Dragomir, M.P.; Pașov, D.; Cocoșilă, L.; Brehar, F.M.; et al. Expression Analysis of Key Epigenetic Regulators in Glioblastoma. In Proceedings of the International Pathology Conference of the Victor Babes Institute, Bucharest, Romania, 2–4 November 2023. [Google Scholar]
- Townson, J.L.; Naumov, G.N.; Chambers, A.F. The role of apoptosis in tumor progression and metastasis. Curr. Mol. Med. 2003, 3, 631–642. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Kim, H.I.; Lee, S.J.; Choi, Y.J.; Kim, M.J.; Kim, T.Y.; Ko, S.G. Quercetin Induces Apoptosis in Glioblastoma Cells by Suppressing Axl/IL-6/STAT3 Signaling Pathway. Am. J. Chin. Med. 2021, 49, 767–784. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, e007530. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Qu, G.Z.; Grundy, P.E.; Narayan, A.; Ehrlich, M. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet. Cytogenet. 1999, 109, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Pornthanakasem, W.; Kongruttanachok, N.; Phuangphairoj, C.; Suyarnsestakorn, C.; Sanghangthum, T.; Oonsiri, S.; Ponyeam, W.; Thanasupawat, T.; Matangkasombut, O.; Mutirangura, A. LINE-1 methylation status of endogenous DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 3667–3675. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Emerging, D.C.L. Roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 2016, 36, 50–58. [Google Scholar] [CrossRef]
- Yamagishi, M.; Uchimaru, K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef]
- Kodach, L.L.; Jacobs, R.J.; Heijmans, J.; van Noesel, C.J.; Langers, A.M.; Verspaget, H.W.; Hommes, D.W.; Offerhaus, G.J.; van den Brink, G.R.; Hardwick, J.C. The role of EZH2 and DNA methylation in the silencing of the tumour suppressor RUNX3 in colorectal cancer. Carcinogenesis 2010, 31, 1567–1575. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Mirkin, B.L.; Dwivedi, R.S. Inhibition of DNA methyltransferase reverses cisplatin induced drug resistance in murine neuroblastoma cells. Cancer Detect. Prev. 2005, 29, 456–463. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P.B. Resistance to cis- and carboplatin initiated by epigenetic changes in ovarian cancer patients. Cancer Drug Resist. 2019, 2, 271–296. [Google Scholar] [CrossRef]
- Urup, T.; Gillberg, L.; Kaastrup, K.; Lü, M.J.S.; Michaelsen, S.R.; Andrée Larsen, V.; Christensen, I.J.; Broholm, H.; Lassen, U.; Grønbaek, K.; et al. Angiotensinogen promoter methylation predicts bevacizumab treatment response of patients with recurrent glioblastoma. Mol. Oncol. 2020, 14, 964–973. [Google Scholar] [CrossRef]
- Wang, G.; Lin, X.; Han, H.; Zhang, H.; Li, X.; Feng, M.; Jiang, C. lncRNA H19 promotes glioblastoma multiforme development by activating autophagy by sponging miR-491-5p. Bioengineered 2022, 13, 11440–11455. [Google Scholar] [CrossRef]
- Liang, W.Q.; Zeng, D.; Chen, C.F.; Sun, S.M.; Lu, X.F.; Peng, C.Y.; Lin, H.Y. Long noncoding RNA H19 is a critical oncogenic driver and contributes to epithelial-mesenchymal transition in papillary thyroid carcinoma. Cancer Manag. Res. 2019, 11, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, Y.; Yu, Y.; Zhong, C.; Lang, Q.; Liang, Z.; Lv, C.; Xu, F.; Tian, Y. Long Noncoding RNA H19: A Novel Therapeutic Target Emerging in Oncology Via Regulating Oncogenic Signaling Pathways. Front. Cell Dev. Biol. 2021, 9, 796740. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, B.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 2014, 5, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Huang, X.; Wu, H.; Yin, G.; Shen, L. LncRNA-H19 gene plays a significant role in regulating glioma cell function. Mol. Genet. Genomic Med. 2021, 9, e1480. [Google Scholar] [CrossRef]
- Zhang, H.M.; Cui, M.Y.; Chen, Z.H. EMX2OS targeting IGF2BP1 represses Wilms’ tumour stemness, epithelial-mesenchymal transition and metastasis. J. Genet. 2023, 102, 12. [Google Scholar] [CrossRef]
- Gu, Y.; Feng, C.; Liu, T.; Zhang, B.; Yang, L. The downregulation of lncRNA EMX2OS might independently predict shorter recurrence-free survival of classical papillary thyroid cancer. PLoS ONE 2018, 13, e0209338. [Google Scholar] [CrossRef]
- Davis, I.J.; Hsi, B.L.; Arroyo, J.D.; Vargas, S.O.; Yeh, Y.A.; Motyckova, G.; Valencia, P.; Perez-Atayde, A.R.; Argani, P.; Ladanyi, M.; et al. Cloning of an α-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc. Natl. Acad. Sci. USA 2003, 100, 6051–6056. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes. Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef]
- Ying, L.; Chen, Q.; Wang, Y.; Zhou, Z.; Huang, Y.; Qiu, F. Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol. Biosyst. 2012, 8, 2289–2294. [Google Scholar] [CrossRef]
- Guo, F.; Liu, Y.; Li, Y.; Li, G. Inhibition of ADP-ribosylation factor-like 6 interacting protein 1 suppresses proliferation and reduces tumor cell invasion in CaSki human cervical cancer cells. Mol. Biol. Rep. 2010, 37, 3819–3825. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Chen, L.; Wang, Y.; Jiang, X.; Xia, H.; Zhuang, Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J. Neurooncol 2015, 121, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, M.; Wang, Z.; Han, S.; Tang, X.; Ge, Y.; Zhou, L.; Zhou, C.; Yuan, Q.; Yang, M. Silencing of long noncoding RNA MALAT1 by miR-101 and miR-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J. Biol. Chem. 2015, 290, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The Noncoding RNA MALAT1 Is a Critical Regulator of the Metastasis Phenotype of Lung Cancer Cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Han, Y.; Wu, Z.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Xie, X.; Zhou, Y.; Du, Z. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016, 7, e2123. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, L.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Zhou, Y.; Du, Z. Downregulation of lncRNA-MALAT1 Affects Proliferation and the Expression of Stemness Markers in Glioma Stem Cell Line SHG139S. Cell Mol. Neurobiol. 2016, 36, 1097–1107. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Y.; Li, J.; Lv, M.; Niu, H.; Tian, Y. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by suppressing miR-155 expression and activating FBXW7 function. Am. J. Cancer Res. 2016, 6, 2561–2574. [Google Scholar]
- Voce, D.J.; Bernal, G.M.; Wu, L.; Crawley, C.D.; Zhang, W.; Mansour, N.M.; Cahill, K.E.; Szymura, S.J.; Uppal, A.; Raleigh, D.R.; et al. Temozolomide Treatment Induces lncRNA MALAT1 in an NF-κB and p53 Codependent Manner in Glioblastoma. Cancer Res. 2019, 79, 2536–2548. [Google Scholar] [CrossRef]
- Li, X.; Luo, Y.; Liu, L.; Cui, S.; Chen, W.; Zeng, A.; Shi, Y.; Luo, L. The long noncoding RNA ZFAS1 promotes the progression of glioma by regulating the miR-150-5p/PLP2 axis. J. Cell Physiol. 2020, 235, 2937–2946. [Google Scholar] [CrossRef]
- Gao, K.; Ji, Z.; She, K.; Yang, Q.; Shao, L. Long non-coding RNA ZFAS1 is an unfavourable prognostic factor and promotes glioma cell progression by activation of the Notch signaling pathway. Biomed. Pharmacother. 2017, 87, 555–560. [Google Scholar] [CrossRef]
- Huang, K.; Yue, X.; Zheng, Y.; Zhang, Z.; Cheng, M.; Li, L.; Chen, Z.; Yang, Z.; Bian, E.; Zhao, B. Development and Validation of an Mesenchymal-Related Long Non-Coding RNA Prognostic Model in Glioma. Front. Oncol. 2021, 11, 726745. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Takahashi, M.; Watanuki, M.; Watanabe, M.; Hiraide, S.; Saijo, K.; Komine, K.; Ishioka, C. lncRNA HAR1B has potential to be a predictive marker for pazopanib therapy in patients with sarcoma. Oncol. Lett. 2021, 21, 455. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.; Pucci, P.; Hirst, M.; Chapman, S.; Wang, Y.; Crea, F.; Heath, C.J. HAR1: An insight into lncRNA genetic evolution. Epigenomics 2021, 13, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.M.; Chen, L.; Li, F.; Zhang, R.; Li, Z.Y.; Chen, F.F.; Jiang, X.D. Over-expression of the long non-coding RNA HOTTIP inhibits glioma cell growth by BRE. J. Exp. Clin. Cancer Res. 2016, 35, 162. [Google Scholar] [CrossRef]
- Li, Z.; Li, M.; Xia, P.; Lu, Z. HOTTIP Mediated Therapy Resistance in Glioma Cells Involves Regulation of EMT-Related miR-10b. Front Oncol 2022, 12, 873561. [Google Scholar] [CrossRef]
- Chae, Y.; Roh, J.; Kim, W. The Roles Played by Long Non-Coding RNAs in Glioma Resistance. Int. J. Mol. Sci. 2021, 22, 6834. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; An, G.; Ma, Q. LncRNA TUG1 acts as a tumor suppressor in human glioma by promoting cell apoptosis. Exp. Biol. Med. 2016, 241, 644–649. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, W. Long noncoding RNA MEG3 suppresses the growth of glioma cells by regulating the miR-96-5p/MTSS1 signaling pathway. Mol. Med. Rep. 2019, 20, 4215–4225. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, P.; Liu, J.; Zheng, J.; Liu, Y.; Chen, J.; Xue, Y. Gas5 exerts tumor-suppressive functions in human glioma cells by targeting miR-222. Mol. Ther. 2015, 23, 1899–1911. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Han, L.; Bao, Z.-S.; Wang, Y.-Y.; Chen, L.-Y.; Yan, W.; Yu, S.-Z.; Pu, P.-Y.; Liu, N.; You, Y.-P.; et al. HOTAIR, a cell cycle–associated long noncoding RNA and a strong predictor of survival, is preferentially expressed in classical and mesenchymal glioma. Neuro-Oncol. 2013, 15, 1595–1603. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Early Apoptosis (%) | Late Apoptosis (%) | Total Apoptosis (%) | |
|---|---|---|---|
| NT | 4.2 | 1.6 | 5.8 |
| SAHA (5 μM) | 6.4 | 6.3 | 12.7 |
| 5-Aza C (5 μM) | 8.4 | 5.2 | 13.6 |
| CPt (50 μM) | 5.6 | 7.7 | 13.3 |
| Pxl (1 μM) | 11.2 | 8.6 | 19.8 |
| Ava (20 μg/mL) | 11.6 | 9.5 | 21.1 |
| Qct (50 μM) | 16 | 10.8 | 26.8 |
| SAHA + CPt | 6.9 | 16 | 22.9 |
| SAHA + Pxl | 11.1 | 11.2 | 22.3 |
| SAHA + Ava | 5.6 | 13 | 18.6 |
| SAHA + Qct | 8.7 | 16.5 | 25.2 |
| 5-Aza C + CPt | 10.1 | 11.4 | 21.5 |
| 5-Aza C + Pxl | 5.7 | 9.7 | 15.4 |
| 5-Aza C + Ava | 4.2 | 12 | 16.2 |
| 5-Aza C + Qct | 5.7 | 13.6 | 19.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albulescu, A.; Botezatu, A.; Fudulu, A.; Hotnog, C.M.; Bostan, M.; Mihăilă, M.; Iancu, I.V.; Plesa, A.; Brasoveanu, L. Combined Effect of Conventional Chemotherapy with Epigenetic Modulators on Glioblastoma. Genes 2025, 16, 138. https://doi.org/10.3390/genes16020138
Albulescu A, Botezatu A, Fudulu A, Hotnog CM, Bostan M, Mihăilă M, Iancu IV, Plesa A, Brasoveanu L. Combined Effect of Conventional Chemotherapy with Epigenetic Modulators on Glioblastoma. Genes. 2025; 16(2):138. https://doi.org/10.3390/genes16020138
Chicago/Turabian StyleAlbulescu, Adrian, Anca Botezatu, Alina Fudulu, Camelia Mia Hotnog, Marinela Bostan, Mirela Mihăilă, Iulia Virginia Iancu, Adriana Plesa, and Lorelei Brasoveanu. 2025. "Combined Effect of Conventional Chemotherapy with Epigenetic Modulators on Glioblastoma" Genes 16, no. 2: 138. https://doi.org/10.3390/genes16020138
APA StyleAlbulescu, A., Botezatu, A., Fudulu, A., Hotnog, C. M., Bostan, M., Mihăilă, M., Iancu, I. V., Plesa, A., & Brasoveanu, L. (2025). Combined Effect of Conventional Chemotherapy with Epigenetic Modulators on Glioblastoma. Genes, 16(2), 138. https://doi.org/10.3390/genes16020138