
Co-Expression Analysis of the ZDHHC19 Palmitoyltransferase–miR-4733–miR-596 Putative Regulatory Axis in Sepsis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. DNA Sampling and Purification
2.3. Blood Sampling, RNA Extraction and cDNA Synthesis
2.4. Human Tissue RNAs
2.5. Quantitative Real-Time PCR Assays
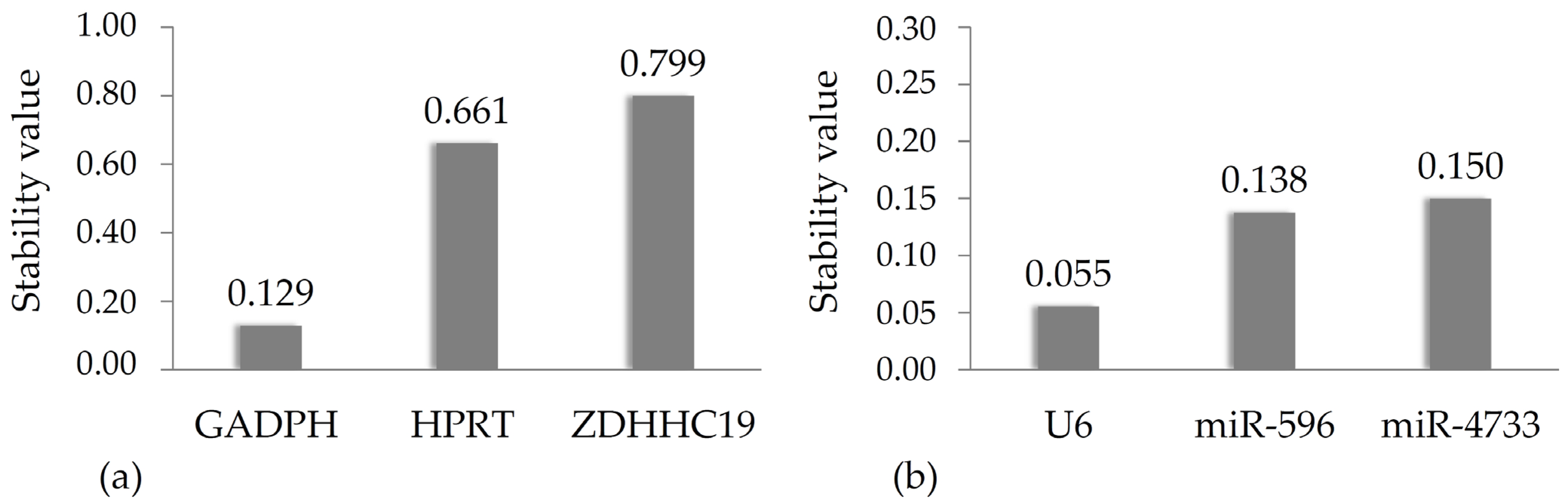
2.6. NormFinder Analysis
2.7. Statistical Analysis
3. Results
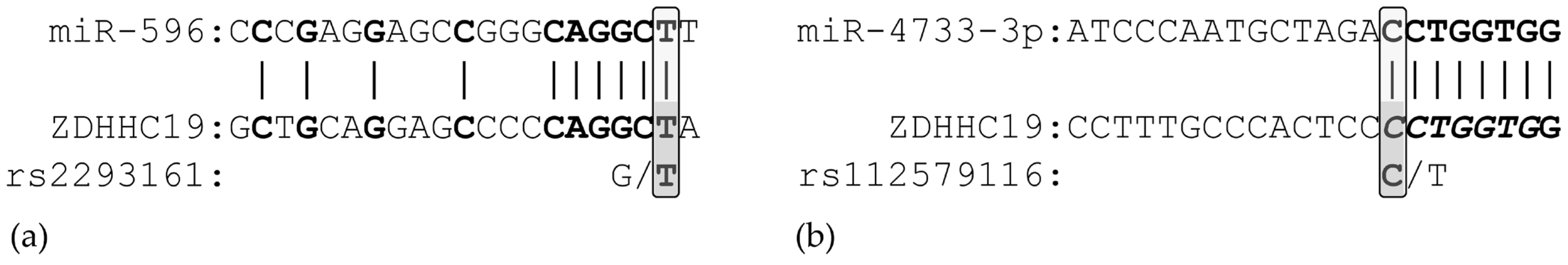
3.1. In Silico Analysis of miRNA Binding
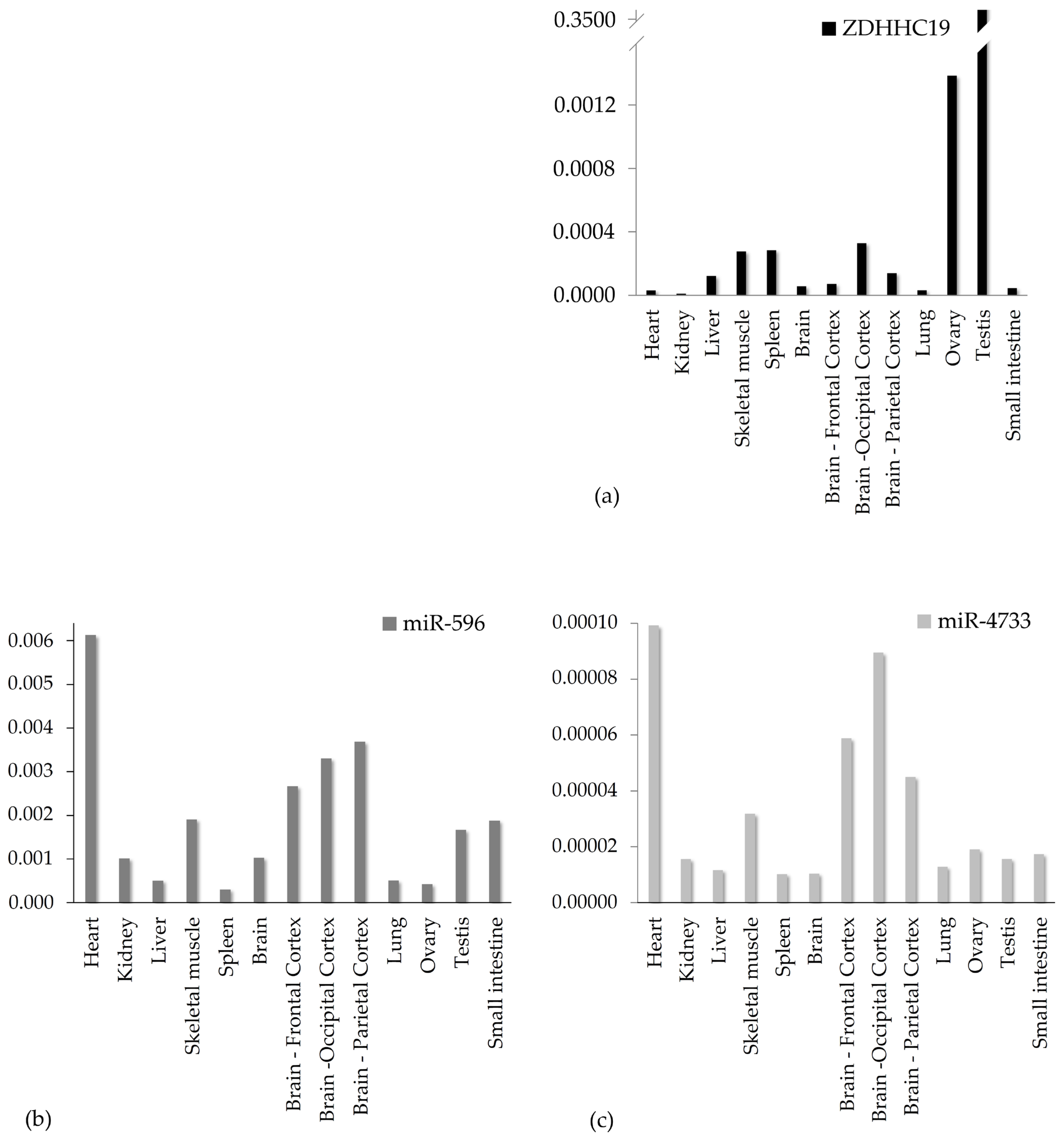
3.2. Gene Expression Analysis in Healthy Human Tissue Samples
3.3. Characterization of Patient Cohorts by Clinical Parameters
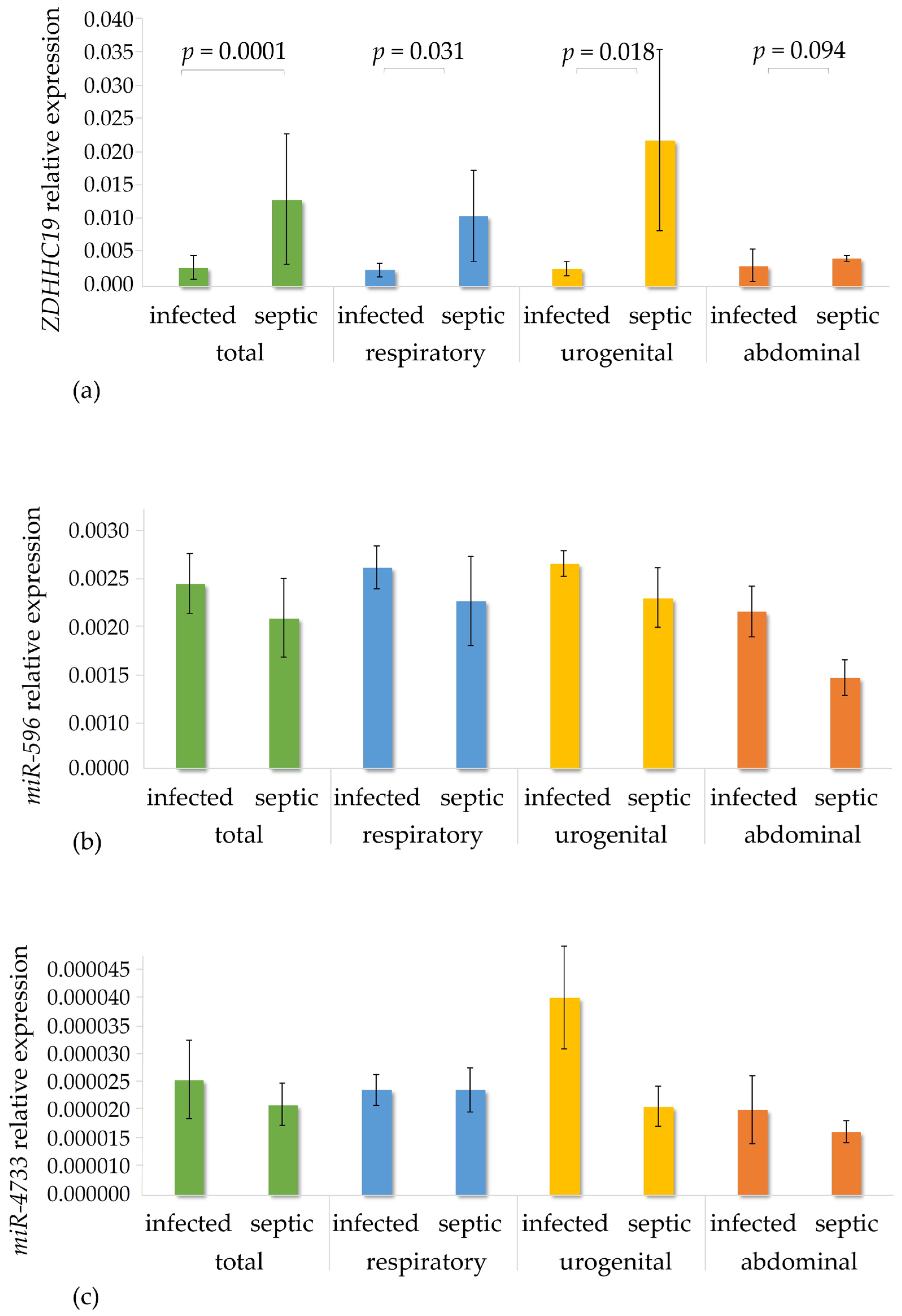
3.4. Gene Expression Analysis in the Patient Cohorts
3.5. Genotyping and Association Analyses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Marik, P.E.; Taeb, A.M. SIRS, qSOFA and new sepsis definition. J. Thorac. Dis. 2017, 9, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Reyes, M.; Filbin, M.R.; Bhattacharyya, R.P.; Billman, K.; Eisenhaure, T.; Hung, D.T.; Levy, B.D.; Baron, R.M.; Blainey, P.C.; Goldberg, M.B.; et al. An immune-cell signature of bacterial sepsis. Nat. Med. 2020, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro da Silva, F.; Cataldi, T.R.; de Lima, T.M.; Starzynski, P.N.; Barbeiro, H.V.; Labate, M.T.; CéMachado, M.C.; de Souza, H.P.; Labate, C.A. Proteomic profiling identifies N-acetylmuramoyl-l-alanine amidase as a novel biomarker of sepsis. Biomark. Med. 2016, 10, 1225–1229. [Google Scholar] [CrossRef]
- Li, L.; Huang, L.; Huang, C.; Xu, J.; Huang, Y.; Luo, H.; Lu, X.; He, S.; Yuan, G.; Chen, L.; et al. The multiomics landscape of serum exosomes during the development of sepsis. J. Adv. Res. 2022, 39, 203–223. [Google Scholar] [CrossRef]
- Memar, M.Y.; Baghi, H.B. Presepsin: A promising biomarker for the detection of bacterial infections. Biomed. Pharmacother. 2019, 111, 649–656. [Google Scholar] [CrossRef]
- Angeletti, S.; Spoto, S.; Fogolari, M.; Cortigiani, M.; Fioravanti, M.; De Florio, L.; Curcio, B.; Cavalieri, D.; Costantino, S.; Dicuonzo, G. Diagnostic and prognostic role of procalcitonin (PCT) and MR-pro-Adrenomedullin (MR-proADM) in bacterial infections. APMIS 2015, 123, 740–748. [Google Scholar] [CrossRef]
- Paul, A.; Newbigging, N.S.; Lenin, A.; Gowri, M.; Varghese, J.S.; Nell, A.J.; Abhilash, K.P.P.; Binu, A.J.; Chandiraseharan, V.K.; Iyyadurai, R.; et al. Role of Neutrophil Gelatinase-associated Lipocalin (NGAL) and Other Clinical Parameters as Predictors of Bacterial Sepsis in Patients Presenting to the Emergency Department with Fever. Indian J. Crit. Care Med. 2023, 27, 176–182. [Google Scholar] [CrossRef]
- Song, L.; Jiang, W.; Lin, H.; Yu, J.; Liu, K.; Zheng, R. Post-translational modifications in sepsis-induced organ dysfunction: Mechanisms and implications. Front. Immunol. 2024, 15, 1461051. [Google Scholar] [CrossRef]
- Binnie, A.; Tsang, J.L.Y.; Hu, P.; Carrasqueiro, G.; Castelo-Branco, P.; Dos Santos, C.C. Epigenetics of Sepsis. Crit. Care Med. 2020, 48, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Song, Y.; Li, J.; Li, Y.; Yu, Y.; Wang, X. Potential biomarker for diagnosis and therapy of sepsis: Lactylation. Immun. Inflamm. Dis. 2023, 11, e1042. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Spencer, C.B.; Ortoga, L.; Zhang, H.; Miao, C. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. 2024, 74, 103194. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Yao, Y.; Hu, H.; Wu, J.; Li, J.; Li, L.; Wu, J.; Sun, M.; Deng, Z.; Zhang, Y.; et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. 2023, 14, 457. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Linder, M.E.; Bamji, S.X.; Dickinson, B.C.; van der Goot, F.G. Mechanisms and functions of protein S-acylation. Nat. Rev. Mol. Cell Biol. 2024, 25, 488–509. [Google Scholar] [CrossRef]
- Jin, J.; Zhi, X.; Wang, X.; Meng, D. Protein palmitoylation and its pathophysiological relevance. J. Cell. Physiol. 2021, 236, 3220–3233. [Google Scholar] [CrossRef]
- Kim, Y.C.; Lee, S.E.; Kim, S.K.; Jang, H.D.; Hwang, I.; Jin, S.; Hong, E.B.; Jang, K.S.; Kim, H.S. Toll-like receptor mediated inflammation requires FASN-dependent MYD88 palmitoylation. Nat. Chem. Biol. 2019, 15, 907–916. [Google Scholar] [CrossRef]
- Kang, J.; Wu, J.; Liu, Q.; Jiang, H.; Li, W.; Li, Y.; Li, X.; Ni, C.; Wu, L.; Liu, M.; et al. FASN regulates STING palmitoylation via malonyl-CoA in macrophages to alleviate sepsis-induced liver injury. Biochim. Biophys. Acta. Mol. Basis Dis. 2024, 1870, 167299. [Google Scholar] [CrossRef]
- Zhu, X.X.; Meng, X.Y.; Zhang, A.Y.; Zhao, C.Y.; Chang, C.; Chen, T.X.; Huang, Y.B.; Xu, J.P.; Fu, X.; Cai, W.W.; et al. Vaccarin alleviates septic cardiomyopathy by potentiating NLRP3 palmitoylation and inactivation. Phytomed. Int. J. Phytother. Phytopharm. 2024, 131, 155771. [Google Scholar] [CrossRef]
- Donkel, S.J.; Portilla Fernández, E.; Ahmad, S.; Rivadeneira, F.; van Rooij, F.J.A.; Ikram, M.A.; Leebeek, F.W.G.; de Maat, M.P.M.; Ghanbari, M. Common and Rare Variants Genetic Association Analysis of Circulating Neutrophil Extracellular Traps. Front. Immunol. 2021, 12, 615527. [Google Scholar] [CrossRef]
- Chen, I.C.; Chen, H.H.; Jiang, Y.H.; Hsiao, T.H.; Ko, T.M.; Chao, W.C. Whole transcriptome analysis to explore the impaired immunological features in critically ill elderly patients with sepsis. J. Transl. Med. 2023, 21, 141. [Google Scholar] [CrossRef]
- He, Y.D.; Wohlford, E.M.; Uhle, F.; Buturovic, L.; Liesenfeld, O.; Sweeney, T.E. The Optimization and Biological Significance of a 29-Host-Immune-mRNA Panel for the Diagnosis of Acute Infections and Sepsis. J. Pers. Med. 2021, 11, 735. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Lu, Y.; Gao, S.; Zhang, L. Palmitoylation of SARS-CoV-2 S protein is critical for S-mediated syncytia formation and virus entry. J. Med. Virol. 2022, 94, 342–348. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, H.; Zhang, L. Fatty Acid Synthase Promotes the Palmitoylation of Chikungunya Virus nsP1. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Tong, D.L.; Kempsell, K.E.; Szakmany, T.; Ball, G. Development of a Bioinformatics Framework for Identification and Validation of Genomic Biomarkers and Key Immunopathology Processes and Controllers in Infectious and Non-infectious Severe Inflammatory Response Syndrome. Front. Immunol. 2020, 11, 380. [Google Scholar] [CrossRef]
- Baumgart, F.; Corral-Escariz, M.; Pérez-Gil, J.; Rodríguez-Crespo, I. Palmitoylation of R-Ras by human DHHC19, a palmitoyl transferase with a CaaX box. Biochim. Biophys. Acta 2010, 1798, 592–604. [Google Scholar] [CrossRef]
- Yhang, M.; Zhou, L.; Xu, Y.; Yang, M.; Xu, Y.; Komaniecki, G.P.; Kosciuk, T.; Chen, X.; Lu, X.; Zou, X.; et al. A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 2020, 586, 434–439. [Google Scholar] [CrossRef]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of phosphoinositide 3-kinase γ by Ras. Curr. Biol. CB 2002, 12, 1068–1075. [Google Scholar] [CrossRef]
- Liang, S.; Zhang, X.; Li, J. Zinc finger Asp-His-His-Cys palmitoyl -acyltransferase 19 accelerates tumor progression through wnt/β-catenin pathway and is upregulated by miR-940 in osteosarcoma. Bioengineered 2022, 13, 7367–7379. [Google Scholar] [CrossRef]
- Zhang, H.; Nguyen-Jackson, H.; Panopoulos, A.D.; Li, H.S.; Murray, P.J.; Watowich, S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010, 116, 2462–2471. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Korma, W.; Mihret, A.; Tarekegn, A.; Chang, Y.; Hwang, D.; Tessema, T.S.; Lee, H. Identification of Circulating miR-22-3p and miR-93-5p as Stable Endogenous Control in Tuberculosis Study. Diagnostics 2020, 10, 868. [Google Scholar] [CrossRef]
- Menyhart, O.; Weltz, B.; Győrffy, B. MultipleTesting.com: A tool for life science researchers for multiple hypothesis testing correction. PLoS ONE 2021, 16, e0245824. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [PubMed]
- Rathour, S.; Kumar, S.; Hadda, V.; Bhalla, A.; Sharma, N.; Varma, S. PIRO concept: Staging of sepsis. J. Postgrad. Med. 2015, 61, 235–242. [Google Scholar] [CrossRef]
- Zhang, T.N.; Wen, R.; Yang, Y.H.; Yang, N.; Liu, C.F. Integration of transcriptomic, proteomic, and metabolomic data to identify lncRNA rPvt1 associations in lipopolysaccharide-treated H9C2 cardiomyocytes. Front. Genet. 2023, 14, 1278830. [Google Scholar] [CrossRef]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.M.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef]
- Beaumont, R.; Tang, K.; Gwee, A. The Sensitivity and Specificity of Procalcitonin in Diagnosing Bacterial Sepsis in Neonates. Hosp. Pediatr. 2024, 14, 199–208. [Google Scholar] [CrossRef]
- Langley, R.J.; Wong, H.R. Early Diagnosis of Sepsis: Is an Integrated Omics Approach the Way Forward? Mol. Diagn. Ther. 2017, 21, 525–537. [Google Scholar] [CrossRef]
- Liu, W.; Geng, F.; Yu, L. Long non-coding RNA MALAT1/microRNA 125a axis presents excellent value in discriminating sepsis patients and exhibits positive association with general disease severity, organ injury, inflammation level, and mortality in sepsis patients. J. Clin. Lab. Anal. 2020, 34, e23222. [Google Scholar] [CrossRef]
- Caidengbate, S.; Akama, Y.; Banerjee, A.; Mokmued, K.; Kawamoto, E.; Gaowa, A.; McCullough, L.D.; Shimaoka, M.; Lee, J.; Park, E.J. MicroRNA Profiles in Intestinal Epithelial Cells in a Mouse Model of Sepsis. Cells 2023, 12, 726. [Google Scholar] [CrossRef]
- Abdelaleem, O.O.; Mohammed, S.R.; El Sayed, H.S.; Hussein, S.K.; Ali, D.Y.; Abdelwahed, M.Y.; Gaber, S.N.; Hemeda, N.F.; El-Hmid, R.G.A. Serum miR-34a-5p and miR-199a-3p as new biomarkers of neonatal sepsis. PLoS ONE 2022, 17, e0262339. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Guo, S. miR-486-5p Serves as a Diagnostic Biomarker for Sepsis and Its Predictive Value for Clinical Outcomes. J. Inflamm. Res. 2021, 14, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Qiu, G.; Ge, M.; Meng, J.; Zhang, G.; Wang, J.; Huang, R.; Shu, Q.; Xu, J. miR-10a in Peripheral Blood Mononuclear Cells Is a Biomarker for Sepsis and Has Anti-Inflammatory Function. Mediat. Inflamm. 2020, 2020, 4370983. [Google Scholar] [CrossRef] [PubMed]
- Buonsenso, D.; Sodero, G.; Valentini, P. Transcript host-RNA signatures to discriminate bacterial and viral infections in febrile children. Pediatr. Res. 2022, 91, 454–463. [Google Scholar] [CrossRef]
- Elek, Z.; Losoncz, E.; Fülep, Z.; Kovács-Nagy, R.; Bánlaki, Z.; Szlobodnyik, G.; Keszler, G.; Rónai, Z. Persistent sepsis-induced transcriptomic signatures in signaling pathways of peripheral blood leukocytes: A pilot study. Hum. Immunol. 2023, 84, 600–608. [Google Scholar] [CrossRef]
- Ma, S.R.; Ma, Q.; Ma, Y.N.; Zhou, W.J. Comprehensive analysis of ceRNA network composed of circRNA, miRNA, and mRNA in septic acute kidney injury patients based on RNA-seq. Front. Genet. 2023, 14, 1209042. [Google Scholar] [CrossRef]
- Luo, X.; Lu, W.; Zhao, J.; Hu, J.; Chen, E.; Fu, S.; Fu, Q. Identification of the TF-miRNA-mRNA co-regulatory networks involved in sepsis. Funct. Integr. Genom. 2022, 22, 481–489. [Google Scholar] [CrossRef]
- Liu, S.M.; Lin, C.H.; Lu, J.; Lin, I.Y.; Tsai, M.S.; Chen, M.H.; Ma, N. miR-596 Modulates Melanoma Growth by Regulating Cell Survival and Death. J. Investig. Dermatol. 2018, 138, 911–921. [Google Scholar] [CrossRef]
- Dai, J.; Yuan, G.; Li, Y.; Zhou, H. MicroRNA-596 is epigenetically inactivated and suppresses prostatic cancer cell growth and migration via regulating Wnt/β-catenin signaling. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2021, 23, 1394–1404. [Google Scholar] [CrossRef]
- Li, C.; Zheng, H.; Xiong, J.; Huang, Y.; Li, H.; Jin, H.; Ai, S.; Wang, Y.; Su, T.; Sun, G.; et al. miR-596-3p suppresses brain metastasis of non-small cell lung cancer by modulating YAP1 and IL-8. Cell Death Dis. 2022, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, J.; Bu, J.; Yang, K.; Xu, S.; Pan, M.; Xiang, D.; Chen, W. MiR-4733-5p promotes gallbladder carcinoma progression via directly targeting kruppel like factor 7. Bioengineered 2022, 13, 10691–10706. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kihara, A.; Sano, T.; Igarashi, Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta 2006, 1761, 474–483. [Google Scholar] [CrossRef]
- Xu, H.; Liu, X.; Ni, H. Clinical significance of miR-19b-3p in patients with sepsis and its regulatory role in the LPS-induced inflammatory response. Eur. J. Med. Res. 2020, 25, 9. [Google Scholar] [CrossRef]
- Zhu, X. MiR-125b but not miR-125a is upregulated and exhibits a trend to correlate with enhanced disease severity, inflammation, and increased mortality in sepsis patients. J. Clin. Lab. Anal. 2020, 34, e23094. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Yue, D.; Zeng, Z.; Yuan, W.; Xu, K. Linkage of lncRNA CRNDE sponging miR-181a-5p with aggravated inflammation underlying sepsis. Innate Immun. 2020, 26, 152–161. [Google Scholar] [CrossRef]
- Zheng, G.; Xiang, W.; Pan, M.; Huang, Y.; Li, Z. Identification of the association between rs41274221 polymorphism in the seed sequence of microRNA-25 and the risk of neonate sepsis. J. Cell. Physiol. 2019, 234, 15147–15155. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.C.; Chen, C.; Zeng, J.; Wang, Q.; Zheng, L.; Yu, H.D. Differential expression of plasma miR-146a in sepsis patients compared with non-sepsis-SIRS patients. Exp. Ther. Med. 2013, 5, 1101–1104. [Google Scholar] [CrossRef]
- Xiang, M.; Zeng, Y.; Yang, R.; Xu, H.; Chen, Z.; Zhong, J.; Xie, H.; Xu, Y.; Zeng, X. U6 is not a suitable endogenous control for the quantification of circulating microRNAs. Biochem. Biophys. Res. Commun. 2014, 454, 210–214. [Google Scholar] [CrossRef]
- Dai, J.; Kumbhare, A.; Youssef, D.; Yao, Z.Q.; McCall, C.E.; El Gazzar, M. Expression of C/EBPβ in myeloid progenitors during sepsis promotes immunosuppression. Mol. Immunol. 2017, 91, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, G.J.; Lang, C.H. Sepsis and AMPK Activation by AICAR Differentially Regulate FoxO-1, -3 and -4 mRNA in Striated Muscle. Int. J. Clin. Exp. Med. 2008, 1, 50–63. [Google Scholar] [PubMed]
- Wang, L.; Fan, H.; Sun, M.; Ye, J.H. SIRT5-mediated HOXA5 desuccinylation inhibits ferroptosis to alleviate sepsis induced-lung injury. Kaohsiung J. Med. Sci. 2025, 41, e12921. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Sequence |
|---|---|
| miR-4733-3p | 5′ CCACCAGGTCTAGCATTGGGAT 3′ |
| miR-596 | 5′ AAGCCTGCCCGGCTCCTCGGG 3′ |
| miR-4293 | 5′ CAGCCTGACAGGAACAG 3′ |
| miR-6078 | 5′ CCGCCTGAGCTAGCTGTGG 3′ |
| U6 | 5′ TTCGTGAAGCGTTCCATATTTT 3′ |
| Infection | Sepsis | p-Value | |
|---|---|---|---|
| Demographics | |||
| Number of patients | 83 | 63 | |
| mean age (years) | 44.2 ± 14.0 | 51.1 ± 12.6 | 5.50 × 10−3 |
| ratio of males (%) | 60.2 | 73.0 | 1.17 × 10−1 |
| Origin of infection | |||
| Abdominal (%) | 26.5 | 4.8 | 1.86 × 10−3 |
| Urogenital (%) | 13.3 | 12.7 | 1.00 × 100 |
| Respiratory (%) | 49.4 | 81.0 | 5.38 × 10−5 |
| Type of infection | |||
| Viral (SARS-CoV-19) (%) | 11.1 | 27.0 | |
| Bacterial (%) | 88.9 | 73.0 | |
| Inflammation | |||
| CRP (mg/L) | 90.5 ± 87.7 | 192.0 ± 127.2 | 1.00 × 10−7 |
| PCT (µg/L) | 1.2 ± 7.2 | 20.5 ± 64.5 | 6.20 × 10−7 |
| Hemogram | |||
| RBC (× 1012/L) | 4.8 ± 0.6 | 4.5 ± 0.9 | 8.81 × 10−2 |
| Hgb (g/L) | 140.1 ± 18.9 | 137.5 ± 19.7 | 3.88 × 10−1 |
| Htc (%) | 23.1 ± 20.6 | 10.3 ± 24.2 | 2.33 × 10−5 |
| WBC (× 109/L) | 11.4 ± 5.7 | 11.7 ± 6.9 | 9.30 × 10−1 |
| neutrophils (%) | 75.4 ± 12.4 | 84.7 ± 8.7 | 1.65 × 10−8 |
| lymphocytes (%) | 15.3 ± 8.1 | 9.9 ± 7.6 | 1.10 × 10−5 |
| PLT (× 109/L) | 267.8 ± 111.6 | 237.8 ± 123.0 | 8.93 × 10−2 |
| Coagulation | |||
| INR | 1.2 ± 0.2 | 1.4 ± 0.3 | 4.19 × 10−3 |
| D-dimer (μg/mL) | 1.7 ± 3.7 | 7.0 ± 14.7 | 8.2 × 10−4 |
| APTT (s) | 35.2 ± 10.1 | 37.2 ± 12.1 | 6.57 × 10−1 |
| fibrinogen (g/L) | 5.5 ± 2.1 | 8.2 ± 5.6 | 1.53 × 10−2 |
| Cardiovascular function | |||
| Systolic pressure (Hgmm) | 128.7 ± 20.0 | 118.2 ± 29.8 | 1.86 × 10−2 |
| Diastolic pressure (Hgmm) | 82.7 ± 11.1 | 71.8 ± 17.7 | 3.24 × 10−4 |
| MAP (Hgmm) | 98.0 ± 13.4 | 90.5 ± 23.7 | 4.05 × 10−2 |
| Heart rate (1/min) | 98.6 ± 17.0 | 105.3 ± 19.2 | 2.57 × 10−2 |
| Shock Index | 0.8 ± 0.2 | 1.0 ± 0.4 | 5.21 × 10−3 |
| Norepinephrin (%) | 0 | 41.7 | - |
| AVP (%) | 0 | 5.3 | - |
| Dobutamine (%) | 0 | 1.7 | - |
| Dopamine (%) | 0 | 3.4 | - |
| Respiratory function | |||
| SpO2 (%) | 96.2 ± 4.2 | 83.1 ± 14.0 | 3.66 × 10−10 |
| Respiratory rate (1/min) | 17.1 ± 3.5 | 28.2 ± 7.2 | 1.55 × 10−15 |
| High flow O2 therapy (%) | 1.2 | 8.6 | - |
| NIV (%) | 0 | 6.9 | - |
| IPPV (%) | 0 | 46.6 | - |
| ECMO (%) | 0 | 5.2 | - |
| Treatment in hospital (days) | 7.9 ± 6.4 | 18.6 ± 19.4 | 9.76 × 10−8 |
| Treatment in ICU (days) | 0.3 ± 1.2 | 6.1 ± 10.7 | 1.28 × 10−7 |
| Mortality (%) | 0 | 23.2 | 6.63 × 10−9 |
| Liver function | |||
| ASAT (IU/L) | 44.4 ± 51.0 | 111.7 ± 367.5 | 8.21 × 10−6 |
| ALAT (IU/L) | 38.9 ± 32.1 | 95.0 ± 304.8 | 5.93 × 10−3 |
| GGT (IU/L) | 87.1 ± 172.3 | 118.6 ± 107.8 | 2.30 × 10−4 |
| ALP (IU/L) | 102.7 ± 141.9 | 102.8 ± 72.2 | 2.47 × 10−1 |
| Total bilirubin (µmol/L) | 9.8 ± 5.6 | 15.6 ± 24.0 | 9.42 × 10−2 |
| LDH (IU/L) | 481.2 ± 828.3 | 526.0 ± 276.0 | 2.64 × 10−3 |
| Renal function | |||
| Crea (µmol/L) | 81.1 ± 28.6 | 138.3 ± 153.0 | 1.45 × 10−3 |
| CN (mmol/L) | 6.4 ± 11.0 | 9.9 ± 10.1 | 4.52 × 10−6 |
| serum Na+ (mmol/L) | 137.9 ± 2.7 | 136.4 ± 5.3 | 1.09 × 10−1 |
| serum K+ (mmol/L) | 4.1 ± 0.6 | 4.2 ± 0.5 | 2.35 × 10−1 |
| IRRT/CRRT (%) | 0 | 17.2 | - |
| Procalcitonin | Neutrophil | Lymphocyte | Platelet | INR | D-Dimer | |
|---|---|---|---|---|---|---|
| Expression of ZDHHC19 | ρ = 0.380 p = 0.029 N = 33 | ρ = 0.449 p = 0.0004 N = 58 | ρ = −0.410 p = 0.0014 N = 58 | ρ = −0.444 p = 0.0005 N = 57 | ρ = 0.421 p = 0.0229 N = 29 | p = 0.1939 |
| Expression of miR-596 | p = 0.1005 | p = 0.0516 | p = 0.0538 | p = 0.1462 | p = 0.2863 | ρ = −0.657 p = 0.0202 N = 12 |
| Expression of miR-4733-3p | p = 0.6287 | p = 0.6840 | p = 0.3909 | p = 0.5584 | p = 0.9182 | p = 0.9656 |
| (A) | ||||||
|---|---|---|---|---|---|---|
| SNP | Allele | Genomic Location (GRCh38.p14) | Intragenic location | MAF | TaqMan ID | HWE (p) |
| rs112579116 | C/T | chr3:196197507 | 3’ UTR | 0.07 (T) | C__99261213_10 | 0.85 |
| rs2293161 | G/T | chr3:196197677 | 3’ UTR | 0.04 (T) | C__15970299_10 | 0.36 |
| (B) | ||||||
| SNP | Allele | Allele Frequency (%) | p-Value | |||
| Infection Cohort | Sepsis Cohort | |||||
| rs112579116 | C | 97.3 | 85 | 0.37 | ||
| T | 2.7 | 3 | ||||
| rs2293161 | G | 95.9 | 80 | 0.19 | ||
| T | 4 | 8 | ||||
| SNP | Infection vs. Sepsis | Sepsis From | ||
|---|---|---|---|---|
| Respiratory | Urogenital | Abdominal | ||
| rs112579116 | 0.7530 | 0.3007 | 0.3126 | 0.8681 |
| rs2293161 | 0.0660 | 0.2380 | 0.8450 | 0.0512 |
| Haplotype rs112579116 rs2293161 | Frequency | Frequency | Chi Square | p | ||
|---|---|---|---|---|---|---|
| 1000 Genomes Project | All Patients | Infected Cohort | Septic Cohort | |||
| GC | 0.873 | 0.914 | 0.882 | 0.932 | 1.789 | 0.1811 |
| GA | 0.044 | 0.057 | 0.084 | 0.041 | 1.952 | 0.1623 |
| AC | 0.083 | 0.027 | 0.027 | 0.027 | 0.000 | 0.9921 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maricza, K.; Elek, Z.; Losoncz, E.; Molnár, K.; Fülep, Z.; Kovács-Nagy, R.; Bánlaki, Z.; Keszler, G.; Rónai, Z. Co-Expression Analysis of the ZDHHC19 Palmitoyltransferase–miR-4733–miR-596 Putative Regulatory Axis in Sepsis. Genes 2025, 16, 359. https://doi.org/10.3390/genes16040359
Maricza K, Elek Z, Losoncz E, Molnár K, Fülep Z, Kovács-Nagy R, Bánlaki Z, Keszler G, Rónai Z. Co-Expression Analysis of the ZDHHC19 Palmitoyltransferase–miR-4733–miR-596 Putative Regulatory Axis in Sepsis. Genes. 2025; 16(4):359. https://doi.org/10.3390/genes16040359
Chicago/Turabian StyleMaricza, Katalin, Zsuzsanna Elek, Eszter Losoncz, Krisztina Molnár, Zoltán Fülep, Réka Kovács-Nagy, Zsófia Bánlaki, Gergely Keszler, and Zsolt Rónai. 2025. "Co-Expression Analysis of the ZDHHC19 Palmitoyltransferase–miR-4733–miR-596 Putative Regulatory Axis in Sepsis" Genes 16, no. 4: 359. https://doi.org/10.3390/genes16040359
APA StyleMaricza, K., Elek, Z., Losoncz, E., Molnár, K., Fülep, Z., Kovács-Nagy, R., Bánlaki, Z., Keszler, G., & Rónai, Z. (2025). Co-Expression Analysis of the ZDHHC19 Palmitoyltransferase–miR-4733–miR-596 Putative Regulatory Axis in Sepsis. Genes, 16(4), 359. https://doi.org/10.3390/genes16040359