
Loose Anagen Hair Associated with Wooly Hair Caused by a Heterozygous, Intronic KRT71 Variant

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Information
2.2. Biologic Samples
2.3. Next-Generation Sequencing (NGS) and Variant Annotation
2.4. Minigene Assay
2.5. Sanger Sequencing
3. Results
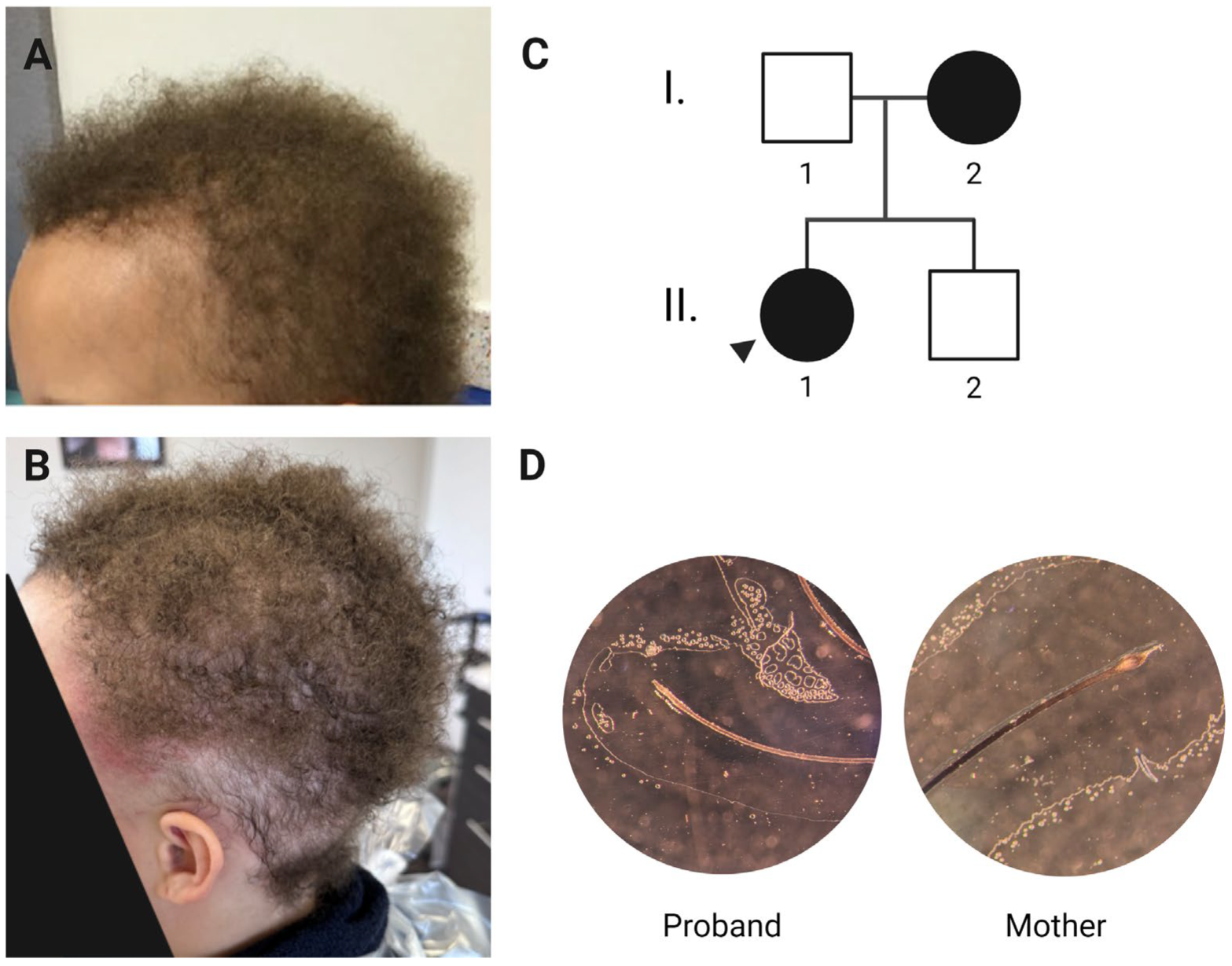
3.1. Clinical History and Phenotype
3.2. Variant Identification and Analysis
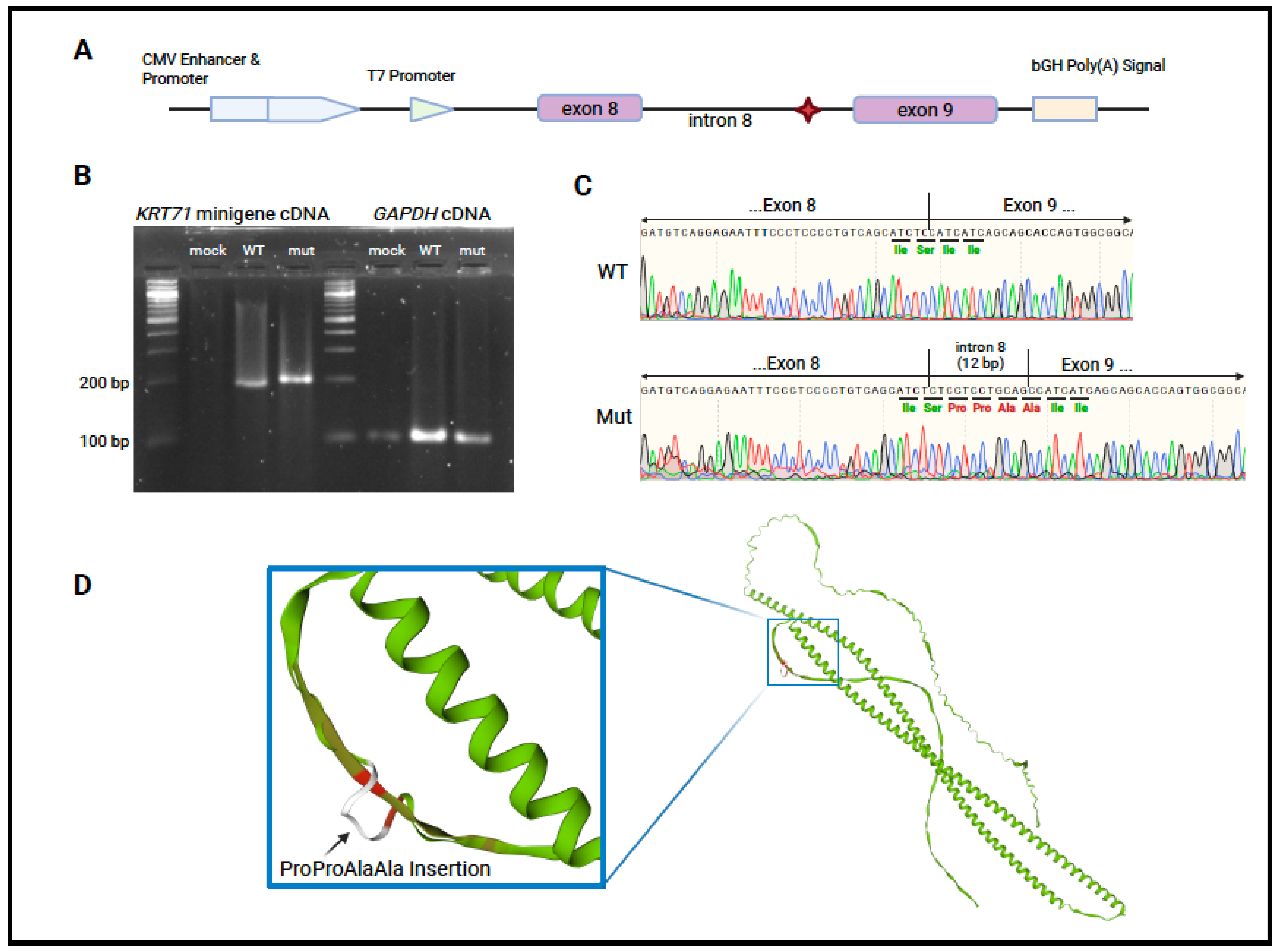
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LAHS | Loose Anagen Hair Syndrome |
| SAHS | Short Anagen Hair Syndrome |
| ORS | Outer Root Sheath |
| IRS | Inner Root Sheath |
| ADWH | Autosomal Dominant Wooly Hair |
| KIF | Keratin Intermediate Filament |
References
- Hamm, H.; Traupe, H. Loose anagen hair of childhood: The phenomenon of easily pluckable hair. J. Am. Acad. Dermatol. 1989, 20, 242–248. [Google Scholar] [CrossRef]
- Price, V.H.; Gummer, C.L. Loose anagen syndrome. J. Am. Acad. Dermatol. 1989, 20, 249–256. [Google Scholar] [CrossRef]
- Swink, S.M.; Castelo-Soccio, L. Loose Anagen Syndrome: A Retrospective Chart Review of 37 Cases. Pediatr. Dermatol. 2016, 33, 507–510. [Google Scholar] [CrossRef]
- Cantatore-Francis, J.L.; Orlow, S.J. Practical guidelines for evaluation of loose anagen hair syndrome. Arch. Dermatol. 2009, 145, 1123–1128. [Google Scholar] [CrossRef]
- Mazzanti, L.; Cacciari, E.; Cicognani, A.; Bergamaschi, R.; Scarano, E.; Forabosco, A. Noonan-like syndrome with loose anagen hair: A new syndrome? Am. J. Med. Genet. A 2003, 118A, 279–286. [Google Scholar] [CrossRef]
- Garcia-Garcia, S.C.; Herz-Ruelas, M.E.; Gomez-Flores, M.; Vazquez-Herrera, N.E.; Misciali, C.; Tosti, A.; Chavez-Alvarez, S.; Ocampo-Candiani, J.; Villarreal-Martinez, A. Association of Trichorhinophalangeal Syndrome and Loose Anagen Syndrome: A Case Report. Ski. Appendage Disord. 2020, 6, 162–167. [Google Scholar] [CrossRef]
- Lee, A.J.; Maino, K.L.; Cohen, B.; Sperling, L. A girl with loose anagen hair syndrome and uncombable, spun-glass hair. Pediatr. Dermatol. 2005, 22, 230–233. [Google Scholar] [CrossRef]
- Sinclair, R.; Cargnello, J.; Chow, C.W. Loose anagen syndrome. Exp. Dermatol. 1999, 8, 297–298. [Google Scholar]
- Chapalain, V.; Winter, H.; Langbein, L.; Le Roy, J.M.; Labreze, C.; Nikolic, M.; Schweizer, J.; Taieb, A. Is the loose anagen hair syndrome a keratin disorder? A clinical and molecular study. Arch. Dermatol. 2002, 138, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.; Yang, J.S.; Castelo-Soccio, L. Bullying and Quality of Life in Pediatric Alopecia Areata. Ski. Appendage Disord. 2017, 3, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Randolph, M.J.; Gwillim, E.C.; Nguyen, B.; Tosti, A. The psychologic impact of loose anagen syndrome and short anagen syndrome. Pediatr. Dermatol. 2022, 39, 4. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Cabezas, A.; Ng, J.C.F.; Canales, J.; Costas, M.J.; Ribeiro, J.M.; Rodrigues, J.R.; McAleer, M.A.; Castelo-Soccio, L.; Simpson, M.A.; et al. Autosomal recessive hypotrichosis with loose anagen hairs associated with TKFC mutations. Br. J. Dermatol. 2021, 184, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Meunier, B.; Mestek-Boukhibar, L.; van den Broek, F.; Maldonado, E.M.; Clement, E.; Weghuber, D.; Spenger, J.; Jaros, Z.; Taha, F.; et al. Bi-allelic Variants in TKFC Encoding Triokinase/FMN Cyclase Are Associated with Cataracts and Multisystem Disease. Am. J. Hum. Genet. 2020, 106, 256–263. [Google Scholar] [CrossRef]
- Mirmirani, P.; Uno, H.; Price, V.H. Abnormal inner root sheath of the hair follicle in the loose anagen hair syndrome: An ultrastructural study. J. Am. Acad. Dermatol. 2011, 64, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Wajid, M.; Petukhova, L.; Kurban, M.; Christiano, A.M. Autosomal-dominant woolly hair resulting from disruption of keratin 74 (KRT74), a potential determinant of human hair texture. Am. J. Hum. Genet. 2010, 86, 632–638. [Google Scholar] [CrossRef]
- Fujimoto, A.; Farooq, M.; Fujikawa, H.; Inoue, A.; Ohyama, M.; Ehama, R.; Nakanishi, J.; Hagihara, M.; Iwabuchi, T.; Aoki, J.; et al. A missense mutation within the helix initiation motif of the keratin K71 gene underlies autosomal dominant woolly hair/hypotrichosis. J. Investig. Dermatol. 2012, 132, 2342–2349. [Google Scholar] [CrossRef]
- Garcia-Hernandez, M.J.; Price, V.H.; Camacho, F.M. Woolly hair associated with loose anagen hair. Acta Derm. Venereol. 2000, 80, 388–389. [Google Scholar]
- Arshdeep, M.R.; De, D.; Handa, S. Loose anagen hair syndrome with diffuse woolly hair phenotype: A rare association. Indian J. Paediatr. Dermatol. 2016, 17, 142–144. [Google Scholar]
- Langbein, L.; Rogers, M.A.; Praetzel, S.; Aoki, N.; Winter, H.; Schweizer, J. A novel epithelial keratin, hK6irs1, is expressed differentially in all layers of the inner root sheath, including specialized huxley cells (Flugelzellen) of the human hair follicle. J. Investig. Dermatol. 2002, 118, 789–799. [Google Scholar] [CrossRef]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An accessible system for testing SNV novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [PubMed]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Roner, S.; Kircher, M. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024, 52, D1143–D1154. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Feng, L.; Wang, J.; Xu, C.; Su, X.; Zhang, G.; Lu, S. A novel PKHD1 splicing variant identified in a fetus with autosomal recessive polycystic kidney disease. Front. Genet. 2023, 14, 1207772. [Google Scholar] [CrossRef]
- Albuisson, J.; Murthy, S.E.; Bandell, M.; Coste, B.; Louis-Dit-Picard, H.; Mathur, J.; Feneant-Thibault, M.; Tertian, G.; de Jaureguiberry, J.P.; Syfuss, P.Y.; et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 2013, 4, 1884. [Google Scholar] [CrossRef]
- Andolfo, I.; Alper, S.L.; De Franceschi, L.; Auriemma, C.; Russo, R.; De Falco, L.; Vallefuoco, F.; Esposito, M.R.; Vandorpe, D.H.; Shmukler, B.E.; et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 2013, 121, 3925–3935. [Google Scholar] [CrossRef]
- Oppermann, H.; Marcos-Graneda, E.; Weiss, L.A.; Gurnett, C.A.; Jelsig, A.M.; Vineke, S.H.; Isidor, B.; Mercier, S.; Magnussen, K.; Zacher, P.; et al. CUX1-related neurodevelopmental disorder: Deep insights into phenotype-genotype spectrum and underlying pathology. Eur. J. Hum. Genet. 2023, 31, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Cogne, B.; Hague, J.; Marcelis, C.L.; Mitter, D.; Oberndorff, K.; Park, S.M.; Ploos van Amstel, H.K.; Simonic, I.; van der Smagt, J.J.; et al. Haploinsufficiency of CUX1 Causes Nonsyndromic Global Developmental Delay with Possible Catch-up Development. Ann. Neurol. 2018, 84, 200–207. [Google Scholar] [CrossRef]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef]
- Richer, J.; Hill, H.L.; Wang, Y.; Yang, M.L.; Hunker, K.L.; Lane, J.; Blackburn, S.; Coleman, D.M.; Eliason, J.; Sillon, G.; et al. A Novel Recurrent COL5A1 Genetic Variant Is Associated with a Dysplasia-Associated Arterial Disease Exhibiting Dissections and Fibromuscular Dysplasia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2686–2699. [Google Scholar] [CrossRef]
- Boisson, B.; Wang, Y.D.; Bosompem, A.; Ma, C.S.; Lim, A.; Kochetkov, T.; Tangye, S.G.; Casanova, J.L.; Conley, M.E. A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(-) B cells. J. Clin. Investig. 2013, 123, 4781–4785. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Eldirany, S.A.; Lomakin, I.B.; Ho, M.; Bunick, C.G. Recent insight into intermediate filament structure. Curr. Opin. Cell Biol. 2021, 68, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.H.; Coulombe, P.A. Defining the properties of the nonhelical tail domain in type II keratin 5: Insight from a bullous disease-causing mutation. Mol. Biol. Cell 2005, 16, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yao, L.; Zhang, X.; Gu, Y.; Yu, H.; Yao, Z.; Zhang, J.; Li, M. Damaged Keratin Filament Network Caused by KRT5 Mutations in Localized Recessive Epidermolysis Bullosa Simplex. Front. Genet. 2021, 12, 736610. [Google Scholar] [CrossRef]
- Jacinto, J.G.P.; Markey, A.D.; Veiga, I.M.B.; Paris, J.M.; Welle, M.; Beever, J.E.; Drogemuller, C. A KRT71 Loss-of-Function Variant Results in Inner Root Sheath Dysplasia and Recessive Congenital Hypotrichosis of Hereford Cattle. Genes 2021, 12, 1038. [Google Scholar] [CrossRef] [PubMed]
- Romero-Benavente, A.; Briano-Rodriguez, C.; Dutra-Quintela, F. Hypotrichosis congenita (KRT71 mutation) in Hereford cattle in Uruguay. Pesqui. Vet. Brasil 2023, 43, e07327. [Google Scholar] [CrossRef]
- Bauer, A.; Hadji Rasouliha, S.; Brunner, M.T.; Jagannathan, V.; Bucher, I.; Bannoehr, J.; Varjonen, K.; Bond, R.; Bergvall, K.; Welle, M.M.; et al. A second KRT71 allele in curly coated dogs. Anim. Genet. 2019, 50, 97–100. [Google Scholar] [CrossRef]
- Cadieu, E.; Neff, M.W.; Quignon, P.; Walsh, K.; Chase, K.; Parker, H.G.; Vonholdt, B.M.; Rhue, A.; Boyko, A.; Byers, A.; et al. Coat variation in the domestic dog is governed by variants in three genes. Science 2009, 326, 150–153. [Google Scholar] [CrossRef]
- Gandolfi, B.; Alhaddad, H.; Joslin, S.E.; Khan, R.; Filler, S.; Brem, G.; Lyons, L.A. A splice variant in KRT71 is associated with curly coat phenotype of Selkirk Rex cats. Sci. Rep. 2013, 3, 2000. [Google Scholar] [CrossRef]
- Gandolfi, B.; Outerbridge, C.A.; Beresford, L.G.; Myers, J.A.; Pimentel, M.; Alhaddad, H.; Grahn, J.C.; Grahn, R.A.; Lyons, L.A. The naked truth: Sphynx and Devon Rex cat breed mutations in KRT71. Mamm. Genome 2010, 21, 509–515. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillippi, E.; Melo, M.; Messingham, K.N.; El-Shanti, H. Loose Anagen Hair Associated with Wooly Hair Caused by a Heterozygous, Intronic KRT71 Variant. Genes 2025, 16, 459. https://doi.org/10.3390/genes16040459
Phillippi E, Melo M, Messingham KN, El-Shanti H. Loose Anagen Hair Associated with Wooly Hair Caused by a Heterozygous, Intronic KRT71 Variant. Genes. 2025; 16(4):459. https://doi.org/10.3390/genes16040459
Chicago/Turabian StylePhillippi, Elizabeth, Marcelo Melo, Kelly N. Messingham, and Hatem El-Shanti. 2025. "Loose Anagen Hair Associated with Wooly Hair Caused by a Heterozygous, Intronic KRT71 Variant" Genes 16, no. 4: 459. https://doi.org/10.3390/genes16040459
APA StylePhillippi, E., Melo, M., Messingham, K. N., & El-Shanti, H. (2025). Loose Anagen Hair Associated with Wooly Hair Caused by a Heterozygous, Intronic KRT71 Variant. Genes, 16(4), 459. https://doi.org/10.3390/genes16040459