Comparative Transcriptome Profiling of Ileal and Cecal Tissues Between Pekin Ducks and Shaoxing Ducks

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Samples Preparation
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Quality Control and Alignment of Raw Sequencing Data
2.4. Gene Expression Analysis
2.5. Differential Gene Expression Analysis
2.6. GO and KEGG Enrichment Analysis of Differentially Expressed Genes
2.7. Real-Time Quantitative PCR (RT-qPCR) Analysis
3. Results
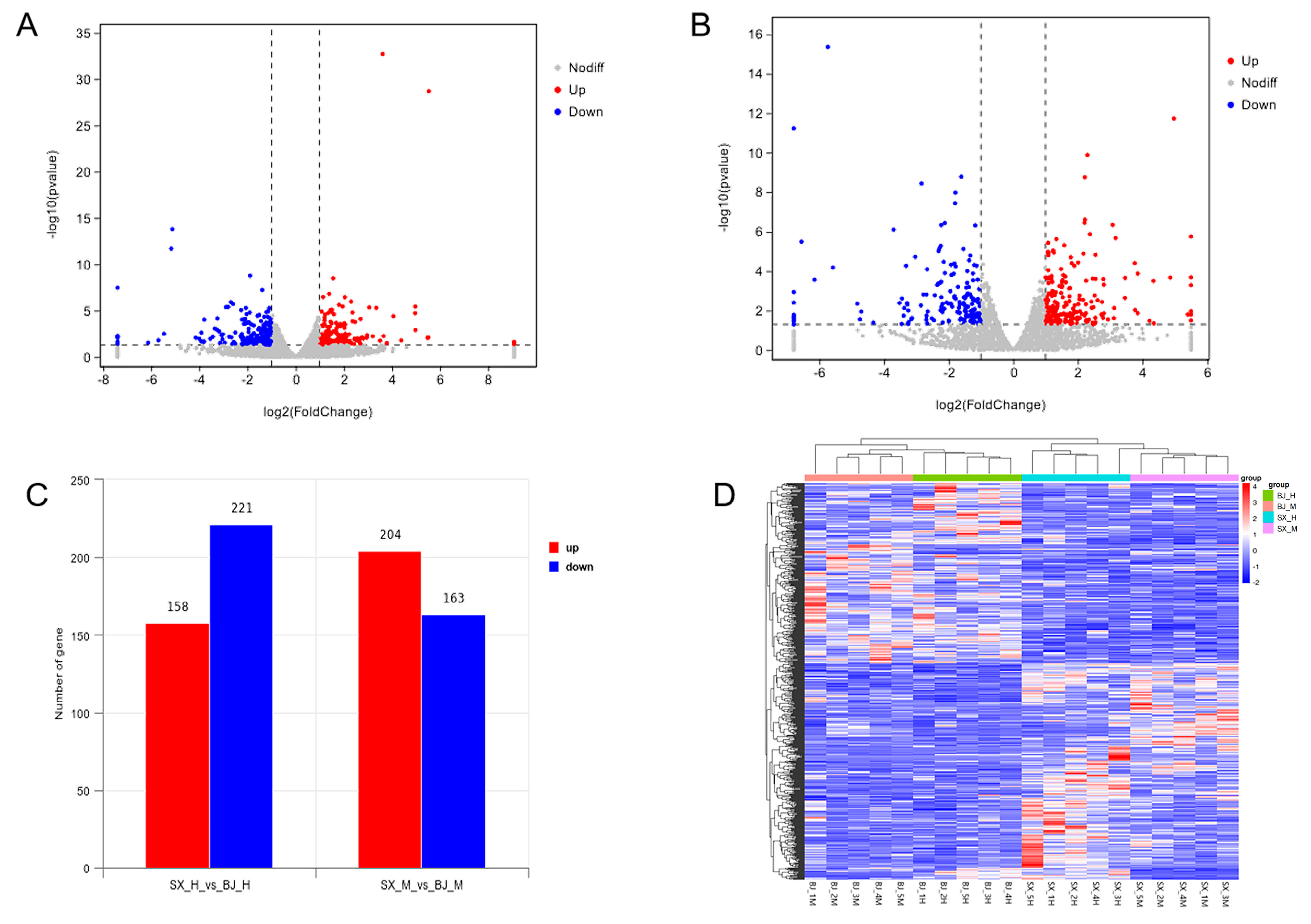
3.1. Transcriptome Sequencing of the Ileum and Cecum from Pekin and Shaoxing Ducks
3.2. Differentially Expressed Genes (DEG) Between Pekin and Shaoxing Ducks
3.3. GO Enrichment Analysis for DEGs
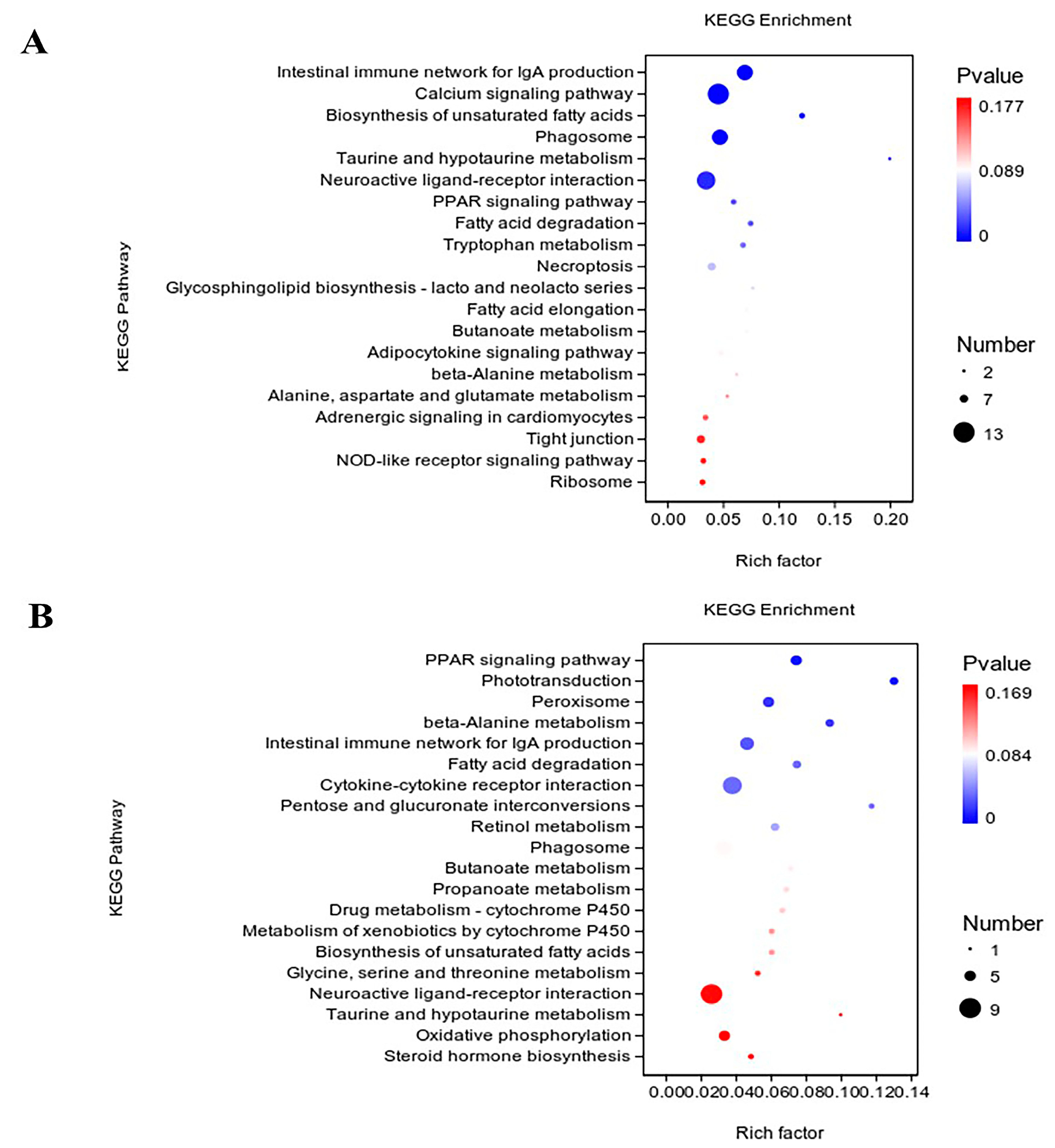
3.4. KEGG Enrichment Analysis for DEGs
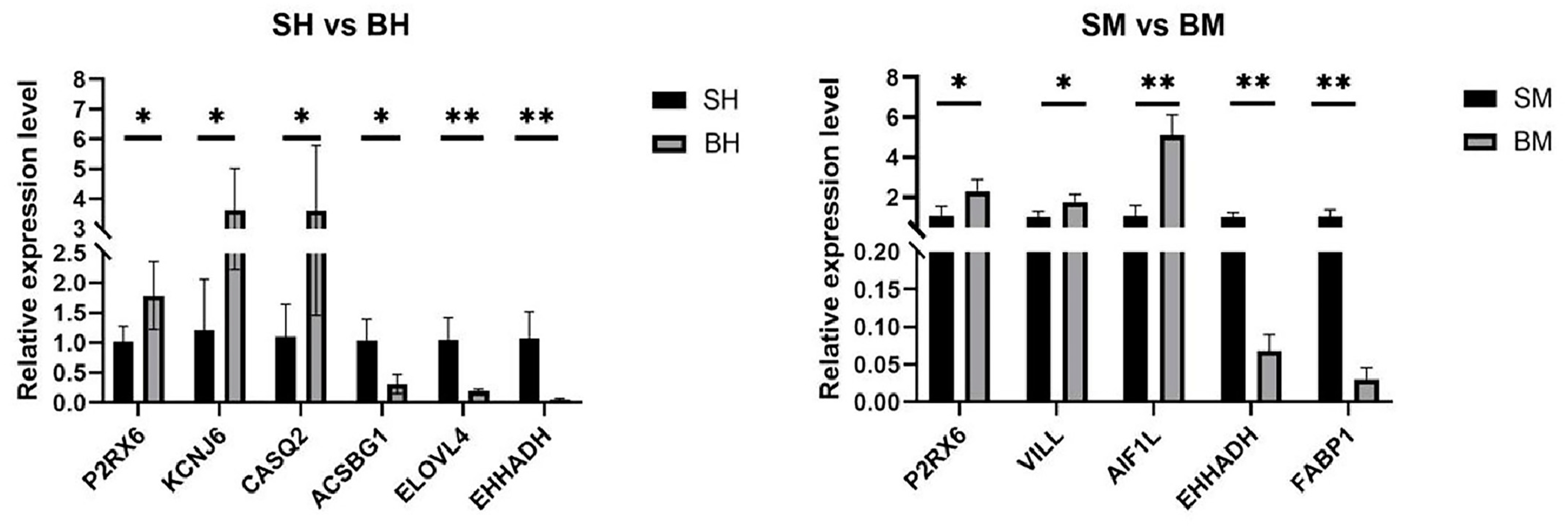
3.5. Verification of DEGs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Jia, Y.; Almeida, P.; Mank, J.E.; van Tuinen, M.; Wang, Q.; Jiang, Z.; Chen, Y.; Zhan, K.; Hou, S.; et al. Whole-Genome Resequencing Reveals Signatures of Selection and Timing of Duck Domestication. GigaScience 2018, 7, giy027. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Yin, Z.-T.; Wang, Z.; Smith, J.; Zhang, F.; Martin, F.; Ogeh, D.; Hincke, M.; Lin, F.-B.; Burt, D.W.; et al. Three Chromosome-Level Duck Genome Assemblies Provide Insights into Genomic Variation during Domestication. Nat. Commun. 2021, 12, 5932. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Z.; Qu, Y.; Li, Q.; Tian, Y.; Chen, L.; Tang, J.; Li, C.; Li, G.; Shen, J.; et al. Genome-Wide Association Studies and Haplotype-Sharing Analysis Targeting the Egg Production Traits in Shaoxing Duck. Front. Genet. 2022, 13, 828884. [Google Scholar] [CrossRef]
- Garro, C.; Brun, A.; Karasov, W.H.; Caviedes-Vidal, E. Small Intestinal Epithelial Permeability to Water-Soluble Nutrients Higher in Passerine Birds than in Rodents. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.Q.; Yang, J.X.; Chen, M.X.; Yan, H.C.; Wang, X.Q. Growth Curves and Age-Related Changes in Carcass Characteristics, Organs, Serum Parameters, and Intestinal Transporter Gene Expression in Domestic Pigeon (Columba Livia). Poult. Sci. 2016, 95, 867–877. [Google Scholar] [CrossRef]
- Nagy, N.; Kovacs, T.; Stavely, R.; Halasy, V.; Soos, A.; Szocs, E.; Hotta, R.; Graham, H.; Goldstein, A.M. Avian Ceca Are Indispensable for Hindgut Enteric Nervous System Development. Development 2021, 148, dev199825. [Google Scholar] [CrossRef]
- Hurwitz, S.; Bar, A.; Katz, M.; Sklan, D.; Budowski, P. Absorption and Secretion of Fatty Acids and Bile Acids in the Intestine of the Laying Fowl. J. Nutr. 1973, 103, 543–547. [Google Scholar] [CrossRef] [PubMed]
- AbuAli, A.M.; Mokhtar, D.M.; Ali, R.A.; Wassif, E.T.; Abdalla, K.E.H. Cellular Elements in the Developing Caecum of Japanese Quail (Coturnix Coturnix Japonica): Morphological, Morphometrical, Immunohistochemical and Electron-Microscopic Studies. Sci. Rep. 2019, 9, 16241. [Google Scholar] [CrossRef]
- Ding, X.; Wang, L.; Liu, H.; Zhang, Y.; Song, W.; Xu, W.; Qiao, W. Comparative Analysis of Growth Models of Pekin Ducks and Shaoxing Ducks. China Poult. 2017, 39, 75–77. [Google Scholar] [CrossRef]
- Wan, Y.; Yang, H.; Han, H.; Zhang, H.; Yang, Y.; Liang, J.; Wang, Z. Study on the Characteristics and Correlation of Early Growth and Development of Digestive Organs of Jiangnan White Goose. China Agric. Sci. Technol. Rev. 2021, 23, 69–77. [Google Scholar] [CrossRef]
- Tian, Y.; Li, G.; Bu, X.; Shen, J.; Tao, Z.; Chen, L.; Zeng, T.; Du, X.; Lu, L. Changes in Morphology and miRNAs Expression in Small Intestines of Shaoxing Ducks in Response to High Temperature. Mol. Biol. Rep. 2019, 46, 3843–3856. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiao, Y.; Yang, H.; Lu, L.; Liu, X.; Lyu, W. Transcriptome Analysis Reveals the Genes Involved in Growth and Metabolism in Muscovy Ducks. BioMed Res. Int. 2021, 6648435. [Google Scholar] [CrossRef]
- Chen, L.; Bao, Y.; Wang, D.; Tian, Y.; Zeng, T.; Gu, T.; Xu, W.; Lu, L. Integrated Omics Analysis Reveals the Differentiation of Intestinal Microbiota and Metabolites between Pekin Ducks and Shaoxing Ducks. Poult. Sci. 2024, 103, 103976. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)). Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nitsan, Z.; Ben-Avraham, G.; Zoref, Z.; Nir, I. Growth and Development of the Digestive Organs and Some Enzymes in Broiler Chicks after Hatching. Br. Poult. Sci. 1991, 32, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-T.; Chou, C.-H.; Wang, C.; Lu, H.-Y.; Yang, W.-Y. Bacillus Amyloliquefaciens and Saccharomyces Cerevisiae Feed Supplements Improve Growth Performance and Gut Mucosal Architecture with Modulations on Cecal Microbiota in Red-Feathered Native Chickens. Anim. Biosci. 2022, 35, 869–883. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, Y.; Ansari, A.R.; Akhtar, M.; Chen, Y.; Cheng, R.; Cui, L.; Nafady, A.A.; Elokil, A.A.; Abdel-Kafy, E.-S.M.; et al. Caecal Microbiota Could Effectively Increase Chicken Growth Performance by Regulating Fat Metabolism. Microb. Biotechnol. 2022, 15, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.; Das, R.; Oak, S.; Mishra, P. Probiotics (Direct-Fed Microbials) in Poultry Nutrition and Their Effects on Nutrient Utilization, Growth and Laying Performance, and Gut Health: A Systematic Review. Animals 2020, 10, 1863. [Google Scholar] [CrossRef]
- Yadav, S.; Jha, R. Strategies to Modulate the Intestinal Microbiota and Their Effects on Nutrient Utilization, Performance, and Health of Poultry. J. Anim. Sci. Biotechnol. 2019, 10, 2. [Google Scholar] [CrossRef]
- Wassie, T.; Cheng, B.; Zhou, T.; Gao, L.; Lu, Z.; Xie, C.; Wu, X. Microbiome-Metabolome Analysis Reveals Alterations in the Composition and Metabolism of Caecal Microbiota and Metabolites with Dietary Enteromorpha Polysaccharide and Yeast Glycoprotein in Chickens. Front. Immunol. 2022, 13, 996897. [Google Scholar] [CrossRef]
- Vinci, M.; Vitello, G.A.; Greco, D.; Treccarichi, S.; Ragalmuto, A.; Musumeci, A.; Fallea, A.; Federico, C.; Calì, F.; Saccone, S.; et al. Next Generation Sequencing and Electromyography Reveal the Involvement of the P2RX6 Gene in Myopathy. Curr. Issues Mol. Biol. 2024, 46, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Mathias, L.S.; Herman-de-Sousa, C.; Cury, S.S.; Nogueira, C.R.; Correia-de-Sá, P.; de Oliveira, M. RNA-Seq Reveals That Anti-Obesity Irisin and Triiodothyronine (T3) Hormones Differentially Affect the Purinergic Signaling Transcriptomics in Differentiated Human Adipocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2023, 1868, 159276. [Google Scholar] [CrossRef]
- Shi, L.; Liu, L.; Ma, Z.; Lv, X.; Li, C.; Xu, L.; Han, B.; Li, Y.; Zhao, F.; Yang, Y.; et al. Identification of Genetic Associations of ECHS1 Gene with Milk Fatty Acid Traits in Dairy Cattle. Anim. Genet. 2019, 50, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhao, X.; Yu, H.; Li, G. Research Note: Identification of Breeding-Related Candidate Genes in Tianjin-Monkey Chickens by Transcriptome Analysis. Poult. Sci. 2023, 102, 102928. [Google Scholar] [CrossRef]
- Xie, S.; Li, M.; Jiang, F.; Yi, Q.; Yang, W. EHHADH is a key gene in fatty acid metabolism pathways in hepatocellular carcinoma: A transcriptomic analysis. Nan Fang Yi Ke Da Xue Xue Bao 2023, 43, 680–693. [Google Scholar] [CrossRef]
- Ranea-Robles, P.; Portman, K.; Bender, A.; Lee, K.; He, J.C.; Mulholland, D.J.; Argmann, C.; Houten, S.M. Peroxisomal L-Bifunctional Protein (EHHADH) Deficiency Causes Male-Specific Kidney Hypertrophy and Proximal Tubular Injury in Mice. Kidney360 2021, 2, 1441–1454. [Google Scholar] [CrossRef]
- Basaki, M.; Asasi, K.; Tabandeh, M.R.; Aminlari, M. Polymorphism Identification and Cardiac Gene Expression Analysis of the Calsequestrin 2 Gene in Broiler Chickens with Sudden Death Syndrome. Br. Poult. Sci. 2016, 57, 151–160. [Google Scholar] [CrossRef]
- Hou, H.; Wang, X.; Yang, C.; Cai, X.; Lv, W.; Tu, Y.; Bao, A.; Wu, Q.; Zhao, W.; Yao, J.; et al. Comparative Genome and Transcriptome Integration Studies Reveal the Mechanism of Pectoral Muscle Development and Function in Pigeons. Front. Genet. 2021, 12, 735795. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Hu, S.; Chen, Q.; Xin, S.; He, Z.; Hu, J.; Hu, B.; He, H.; Liu, H.; Li, L.; et al. Role of SNPs Located in the Exon 9 of ATAPA1 Gene on Goose Egg Production. Poult. Sci. 2023, 102, 102488. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, D.; Li, X.; Ge, M.; Hou, Z. Research Note: Identification of Core Promoter Region of the Polyunsaturated Fatty Acid Synthesis-Related Gene Family in Chicken. Poult. Sci. 2023, 102, 102857. [Google Scholar] [CrossRef]
- Gharib-Naseri, K.; de Las Heras-Saldana, S.; Kheravii, S.; Qin, L.; Wang, J.; Wu, S.-B. Necrotic Enteritis Challenge Regulates Peroxisome Proliferator-1 Activated Receptors Signaling and β-Oxidation Pathways in Broiler Chickens. Anim. Nutr. 2021, 7, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Yasuda-Yamahara, M.; Rogg, M.; Yamahara, K.; Maier, J.I.; Huber, T.B.; Schell, C. AIF1L Regulates Actomyosin Contractility and Filopodial Extensions in Human Podocytes. PLoS ONE 2018, 13, e0200487. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Maita, T.; Miyanishi, T.; Watanabe, S.; Matsuda, G. Amino Acid Sequence of the 203-Residue Fragment of the Heavy Chain of Chicken Gizzard Myosin Containing the SH1-Type Cysteine Residue. J. Biochem. 1986, 100, 1433–1447. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Corsico, B. The Multifunctional Family of Mammalian Fatty Acid-Binding Proteins. Annu. Rev. Nutr. 2023, 43. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Product Size (bp) | TM (°C) | Amplification Efficiency | Primer Sequences (5′ to 3′) |
|---|---|---|---|---|
| GAPDH | 99 | 60 | 100% | F: TCTGGCAAAGTGGAAGTGGT R: CCGGAAGTGGCCATGAGTAG |
| P2RX6 | 78 | 60 | 100% | F: CAAACCAACGACTCCACCTA R: ATGCTGATGCTTCCTCCC |
| KCNJ6 | 82 | 60 | 100% | F: TGACATGCCAAGCTCGAAGT R: TCCTCCAGAGTGAGGACAGG |
| CASQ2 | 98 | 60 | 100% | F: CCAGGAGCATCGGGTTTG R: TTTGGCGAGTTTGGCATC |
| ACSBG1 | 60 | 60 | 100% | F: TCCAGGAAAGCAGCCAAGAG R: TGCTACGCTGTGGAATCGTT |
| EHHADH | 207 | 60 | 100% | F: GCTTGGGCCCCTTAGTTTCT R: CGATCAGTCTGGGTAGTCGC |
| ELOVL4 | 67 | 60 | 100% | F: CTCGAGTTCTACCGCTGGAC R: ACTGCATCAAGGGCCAATCA |
| AIF1L | 61 | 60 | 100% | F: TCTGTGACCCGAAGTTCAGTG R: TCTTTGAACACCGCCAGCTT |
| VILL | 65 | 60 | 100% | F: CAAAACTGCCAAGGTGGAGC R: CTAGCTCTGGTGTCGCATGC |
| FABP1 | 66 | 60 | 100% | F: CTGAAGGGGCTGAAATCCGT R: TCCCATGGTCATTGTGTGGG |
| Samples | Read Numbers | Clean Reads | Clean Bases | Clean Ratio (%) | Q30 (%) |
|---|---|---|---|---|---|
| BJ_1H | 49915050 | 46955144 | 7090226744 | 94.07 | 92.74 |
| BJ_2H | 44497058 | 42069004 | 6352419604 | 94.54 | 92.75 |
| BJ_3H | 44070764 | 41686758 | 6294700458 | 94.59 | 92.18 |
| BJ_4H | 55879402 | 52890360 | 7986444360 | 94.65 | 93.21 |
| BJ_5H | 43124648 | 40771040 | 6156427040 | 94.54 | 92.44 |
| SX_1H | 51437022 | 48631200 | 7343311200 | 94.55 | 92.03 |
| SX_2H | 46469116 | 43974506 | 6640150406 | 94.63 | 92.60 |
| SX_3H | 42176790 | 39892012 | 6023693812 | 94.58 | 93.03 |
| SX_4H | 50527934 | 47843848 | 7224421048 | 94.69 | 92.96 |
| SX_5H | 43820670 | 41437276 | 6257028676 | 94.56 | 93.04 |
| BJ_1M | 45677740 | 43254128 | 6531373328 | 94.69 | 93.00 |
| BJ_2M | 42250402 | 39929594 | 6029368694 | 94.51 | 93.09 |
| BJ_3M | 50843934 | 48039702 | 7253995002 | 94.48 | 92.46 |
| BJ_4M | 46317106 | 43723224 | 6602206824 | 94.40 | 92.18 |
| BJ_5M | 45838652 | 43283748 | 6535845948 | 94.43 | 92.34 |
| SX_1M | 43444586 | 41150334 | 6213700434 | 94.72 | 91.01 |
| SX_2M | 50425364 | 47622128 | 7190941328 | 94.44 | 91.96 |
| SX_3M | 47810534 | 45235762 | 6830600062 | 94.61 | 92.46 |
| SX_4M | 49549788 | 46838862 | 7072668162 | 94.53 | 91.62 |
| SX_5M | 48650494 | 45987318 | 6944085018 | 94.53 | 92.46 |
| Organization | Different Expression Type | Genes | Description | log2FoldChange | p-Value |
|---|---|---|---|---|---|
| Ileum | Up-regulated | P2RX6 | Purinergic receptor P2X, ligand-gated ion channel 6 | 1.620 | 0.017 |
| KCNJ6 | Potassium Inwardly Rectifying Channel Subfamily J Member 6 | 2.241 | 0.019 | ||
| CASQ2 | Calsequestrin 2 | 2.673 | 0.010 | ||
| Down-regulated | EHHADH | Enoyl-CoA Hydratase And 3-Hydroxyacyl CoA Dehydrogenase | 1.152 | 0.003 | |
| ACSBG1 | Acyl-CoA Synthetase Bubblegum Family Member 1 | 1.727 | 0.025 | ||
| ELOVL4 | ELOVL Fatty Acid Elongase 4 | 1.733 | 0.003 | ||
| Cecum | Up-regulated | P2RX6 | Purinergic receptor P2X, ligand-gated ion channel 6 | 1.979 | 0.029 |
| AIF1L | Allograft Inflammatory Factor 1 Like | 1.314 | 0.001 | ||
| VILL | Villin Like | 1.564 | 0.014 | ||
| Down-regulated | EHHADH | Enoyl-CoA Hydratase And 3-Hydroxyacyl CoA Dehydrogenase | 1.789 | 0.000 | |
| FABP1 | Fatty Acid Binding Protein 1 | 1.400 | 0.006 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Hu, Z.; Zhao, A.; Zeng, T.; Gu, T.; Xu, W.; Tian, Y.; Lu, L.; Chen, L. Comparative Transcriptome Profiling of Ileal and Cecal Tissues Between Pekin Ducks and Shaoxing Ducks. Genes 2025, 16, 488. https://doi.org/10.3390/genes16050488
Wang D, Hu Z, Zhao A, Zeng T, Gu T, Xu W, Tian Y, Lu L, Chen L. Comparative Transcriptome Profiling of Ileal and Cecal Tissues Between Pekin Ducks and Shaoxing Ducks. Genes. 2025; 16(5):488. https://doi.org/10.3390/genes16050488
Chicago/Turabian StyleWang, Dandan, Zhengyu Hu, Ayong Zhao, Tao Zeng, Tiantian Gu, Wenwu Xu, Yong Tian, Lizhi Lu, and Li Chen. 2025. "Comparative Transcriptome Profiling of Ileal and Cecal Tissues Between Pekin Ducks and Shaoxing Ducks" Genes 16, no. 5: 488. https://doi.org/10.3390/genes16050488
APA StyleWang, D., Hu, Z., Zhao, A., Zeng, T., Gu, T., Xu, W., Tian, Y., Lu, L., & Chen, L. (2025). Comparative Transcriptome Profiling of Ileal and Cecal Tissues Between Pekin Ducks and Shaoxing Ducks. Genes, 16(5), 488. https://doi.org/10.3390/genes16050488