
Effects of Different Stages of Training on the Intestinal Microbes of Yili Horses Analyzed Using Metagenomics

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Groups
2.2. Sample Collection
2.2.1. Fecal Samples
2.2.2. Sample Results from the 2000 m Test Race
2.3. Sample Quality Control
2.4. Library Construction, Quality Control, and Sequencing
2.5. Bioinformatics Analysis Pipeline
2.6. Statistical Analysis
3. Results
3.1. Test Match Results
3.2. Statistical Results of the Macrogenome Sequencing Data
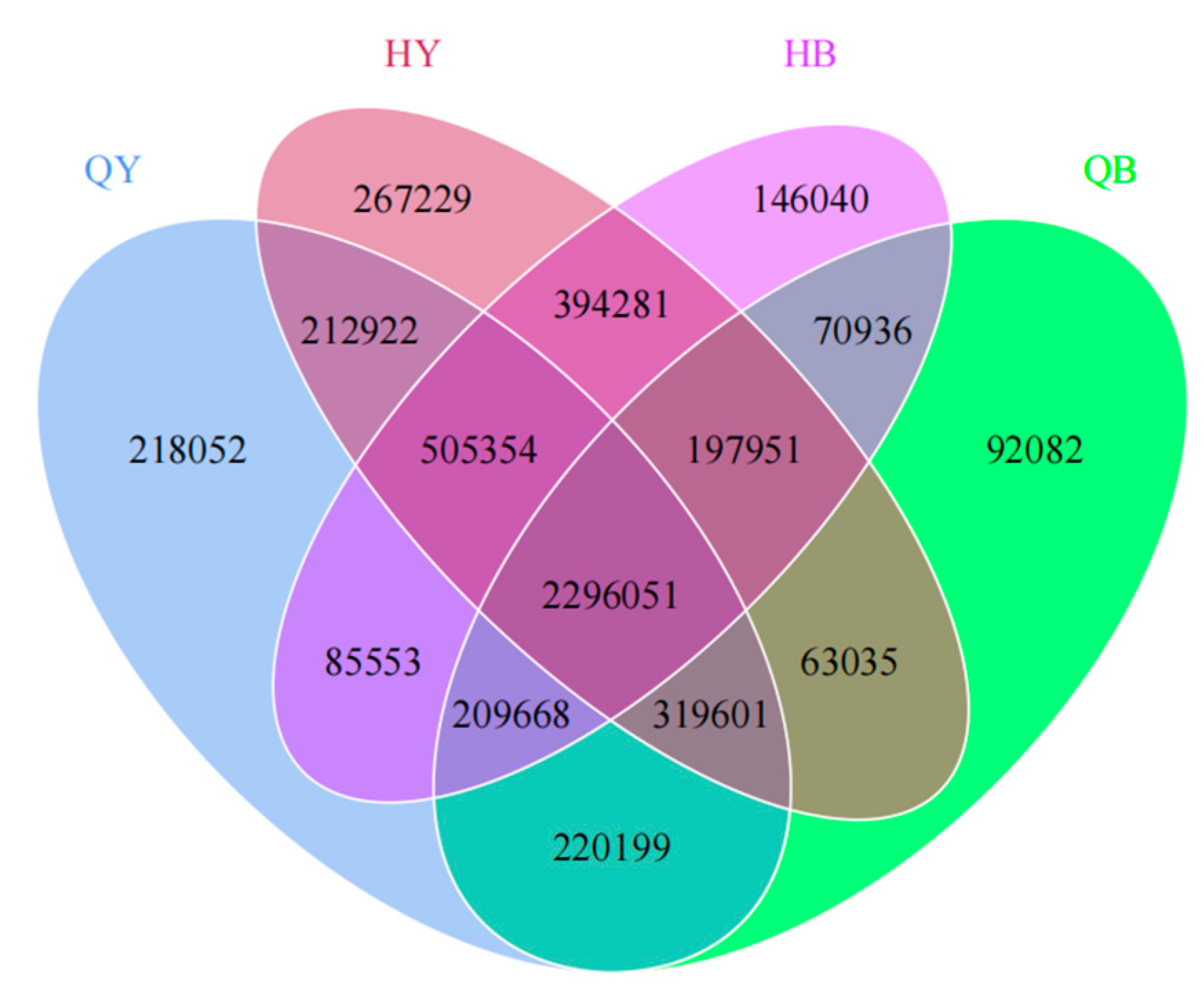
3.3. Analysis of Differences in the Number of Genes
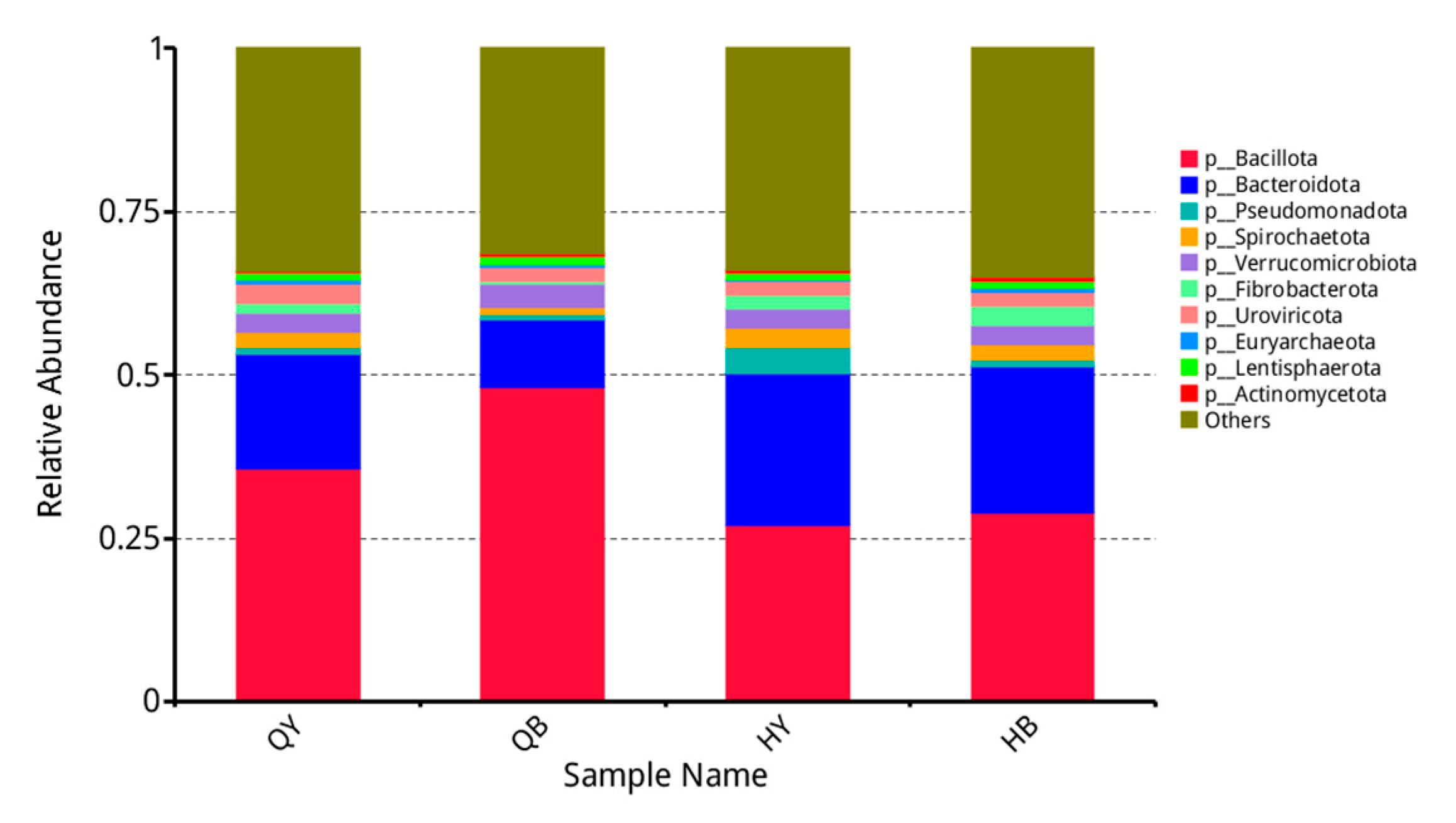
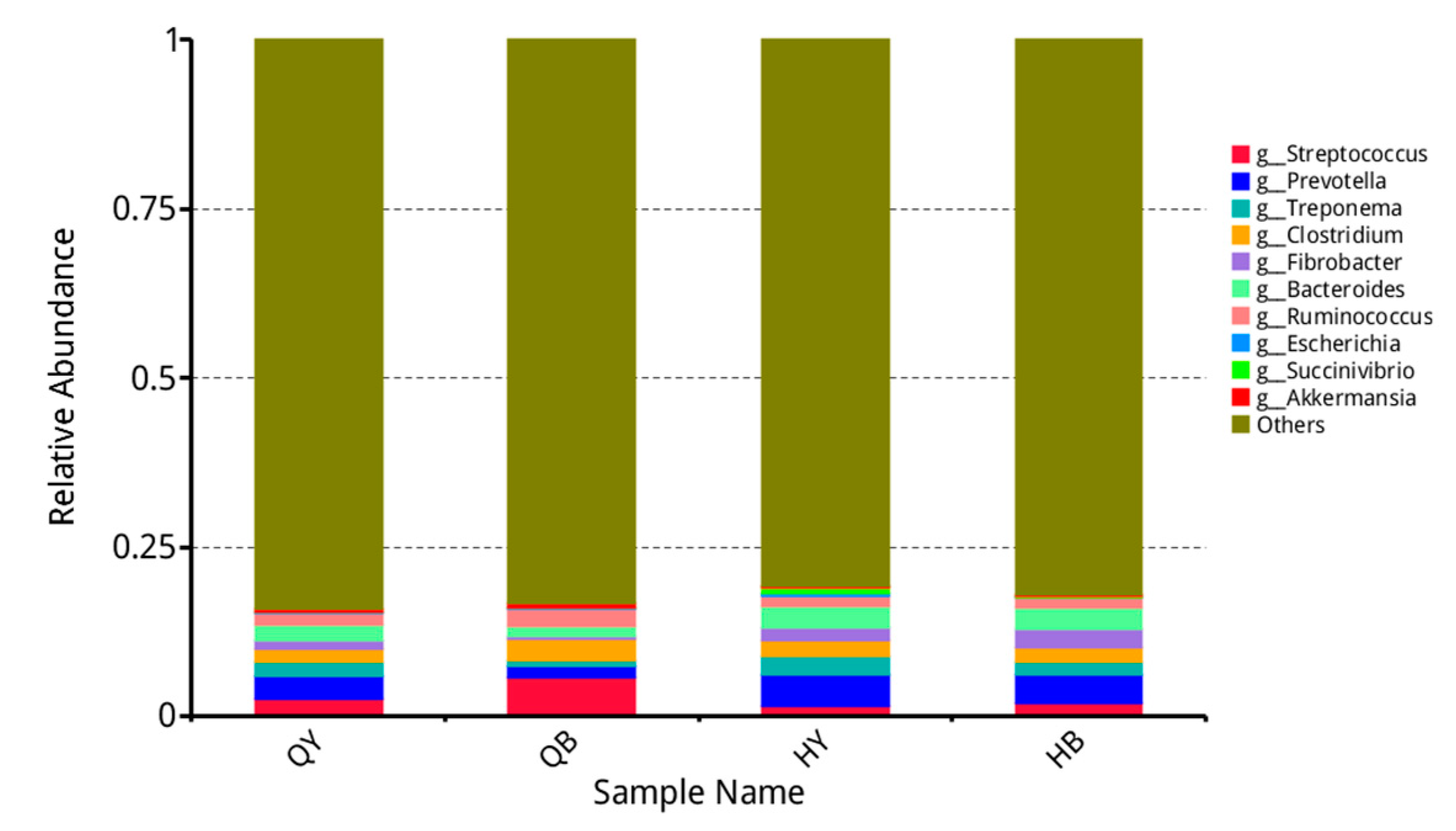
3.4. Analysis of the Relative Abundance of Different Levels of Fecal Flora in Yili Horses
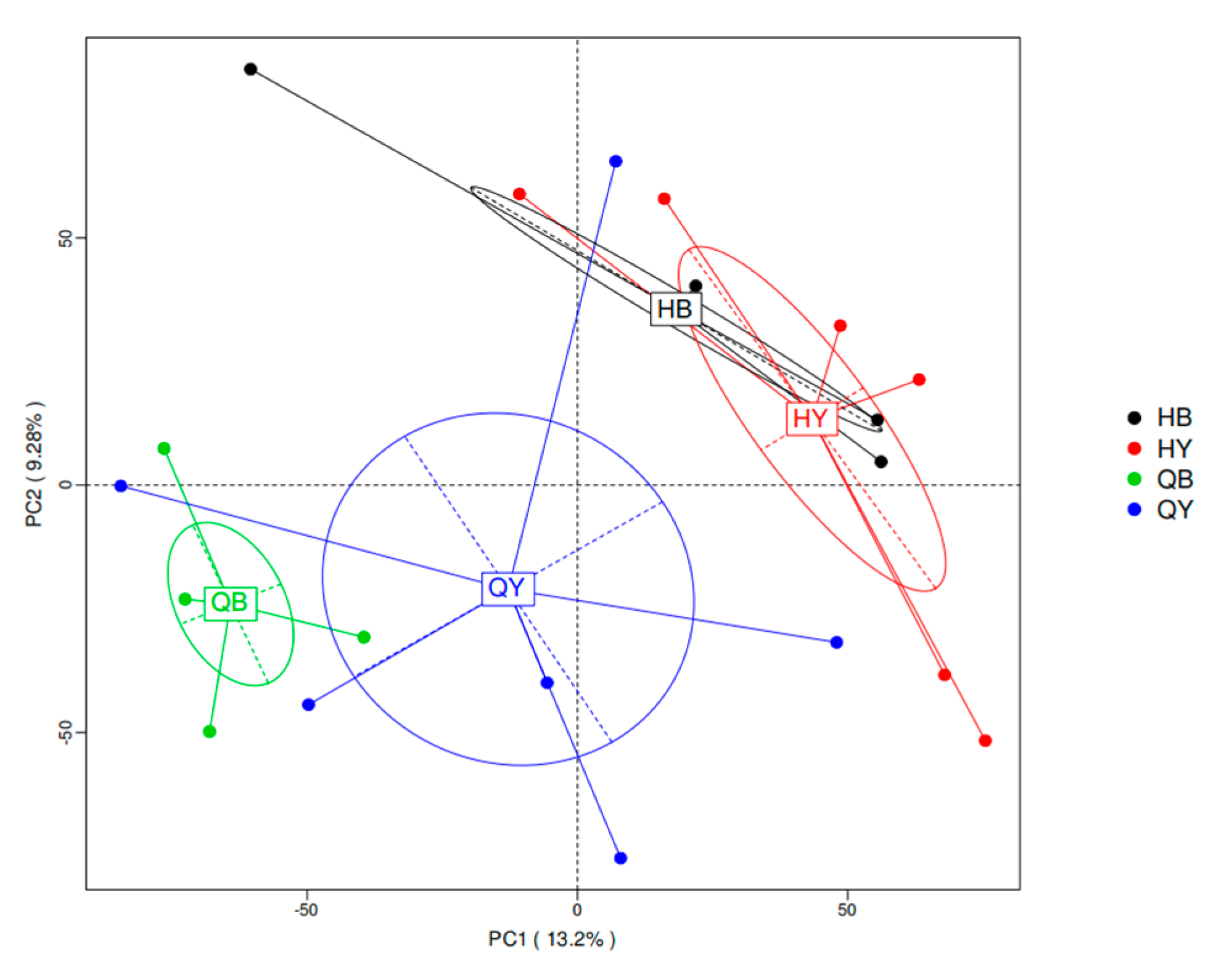
3.5. Principal Component Analysis of Intestinal Microbial Species Abundance in Yili Horses
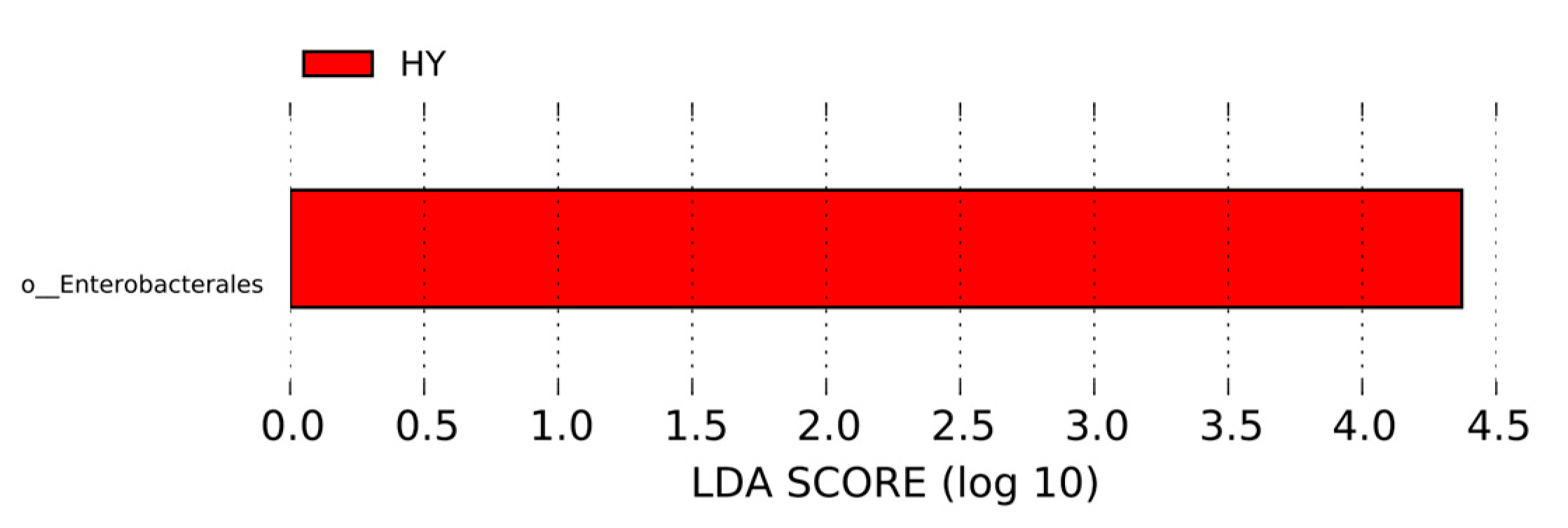
3.6. Linear Discriminant Analysis Effect Size Analysis of Species Differing Between Groups
3.7. Notes on the Intestinal Microbiome Function of Yili Horses
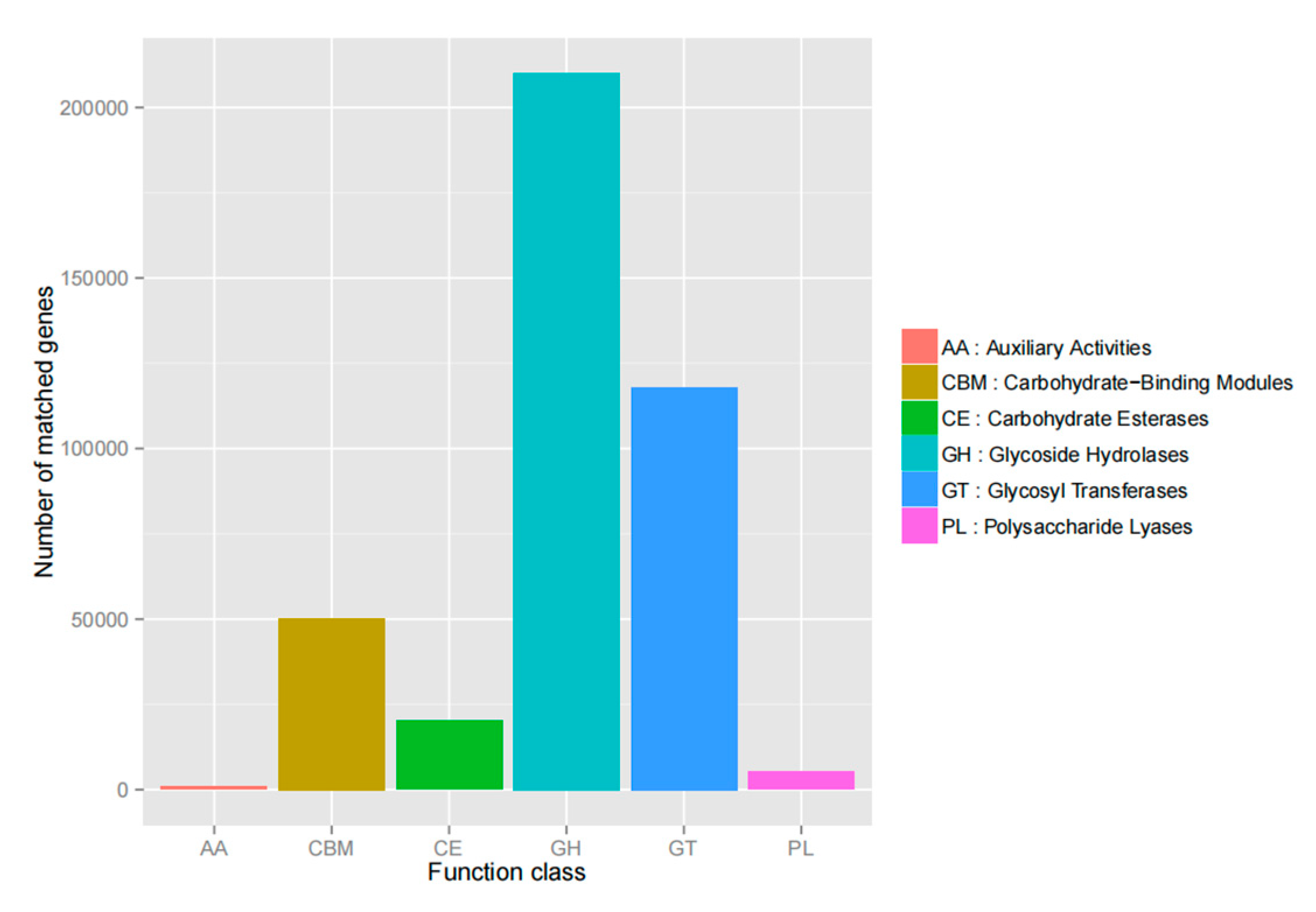
3.7.1. Annotated Gene Number Statistics
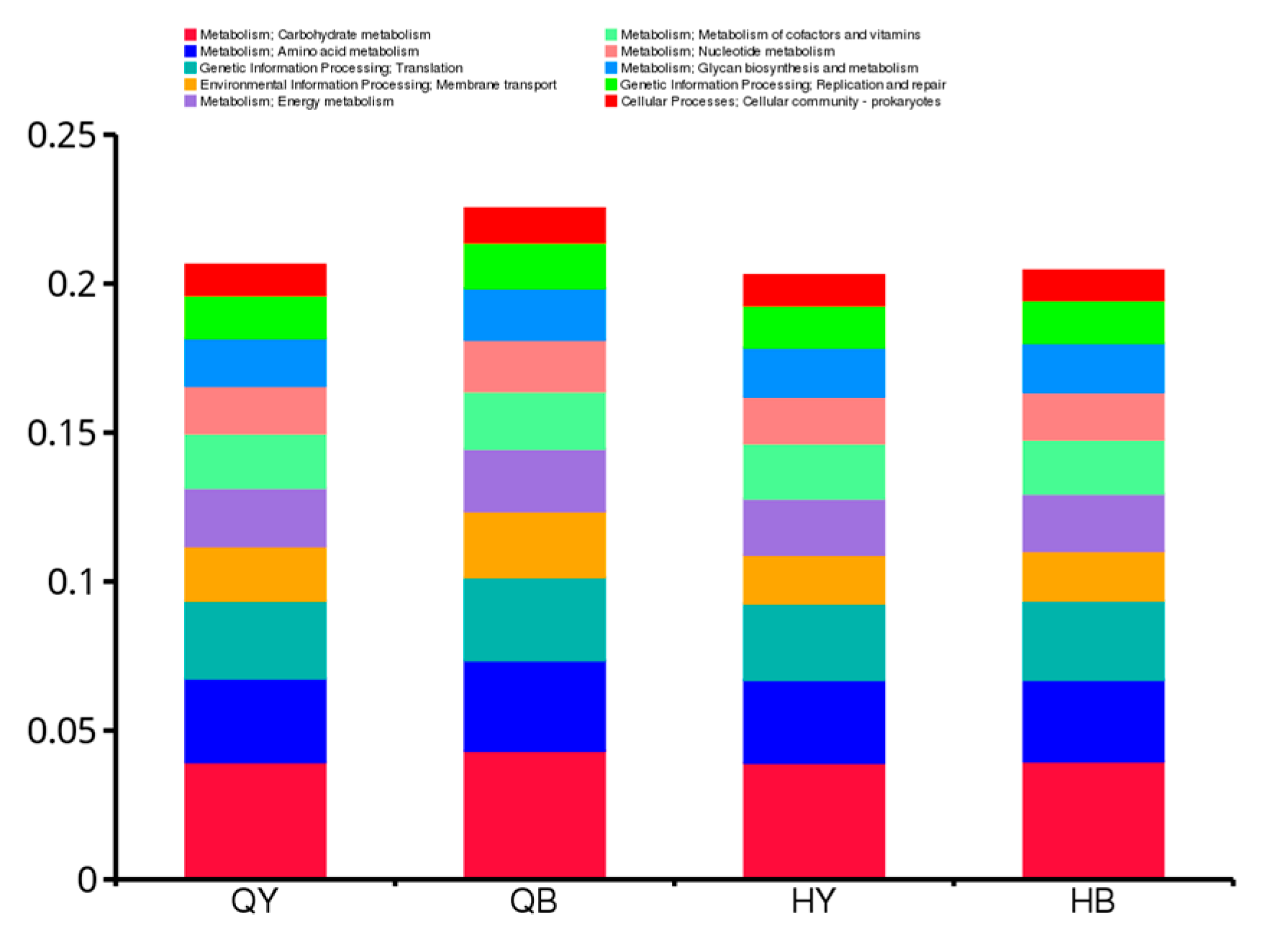
3.7.2. Functional Analysis
3.7.3. Functional Relative Abundance Cluster Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Okamoto, T.; Morino, K.; Ugi, S.; Nakagawa, F.; Lemecha, M.; Ida, S.; Ohashi, N.; Sato, D.; Fujita, Y.; Maegawa, H. Microbiome potentiates endurance exercise through intestinal acetate production. Am. J. Physiol. Metab. 2019, 316, E956–E966. [Google Scholar] [CrossRef]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut 2018, 67, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuño, M.I.; Seoane, L.M.; Murri, M.; Pardo, M.; Gomez-Zumaquero, J.M.; Cardona, F.; Casanueva, F.; Tinahones, F.J. Gut microbiota composition in male rat models under different nutritional status and physical activity and its association with serum leptin and ghrelin levels. PLoS ONE 2013, 8, e65465. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Fernández, M.; Porras, D.; García-Mediavilla, M.V.; Román-Sagüillo, S.; González-Gallego, J.; Nistal, E.; Sánchez-Campos, S. Aging, Gut microbiota and metabolic diseases: management through physical exercise and nutritional interventions. Nutrients 2020, 13, 16. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Al Jassim, R.A.M.; Andrews, F.M. The bacterial community of the horse gastrointestinal tract and its relation to fermentative acidosis, laminitis, colic, and stomach ulcers. Vet. Clin. Equine Pract. 2009, 25, 199–215. [Google Scholar] [CrossRef]
- Mach, N.; Midoux, C.; Leclercq, S.; Pennarun, S.; Le Moyec, L.; Rué, O.; Robert, C.; Sallé, G.; Barrey, E. The first horse gut microbiome gene catalog reveals that rare microbiome ensures better cardiovascular fitness in endurance horses. bioRxiv 2022. [Google Scholar] [CrossRef]
- Dougal, K.; de la Fuente, G.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Newbold, C.J. Identification of a core bacterial community within the large intestine of the horse. PLoS ONE 2013, 8, e77660. [Google Scholar] [CrossRef]
- Messmann, S.; Bagu, E.; Robia, C.; Palme, R. Measurement of glucocorticoid metabolite concentrations in faeces of domestic livestock. J. Veter- Med. Ser. A 1999, 46, 621–631. [Google Scholar] [CrossRef]
- Castejon-Riber, C.; Riber, C.; Rubio, M.D.; Agüera, E.; Muñoz, A. Objectives, Principles, and Methods of Strength Training for Horses. J. Equine Vet. Sci. 2017, 56, 93–103. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef]
- Scheiman, J.; Luber, J.M.; Chavkin, T.A.; MacDonald, T.; Tung, A.; Pham, L.-D.; Wibowo, M.C.; Wurth, R.C.; Punthambaker, S.; Tierney, B.T.; et al. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat. Med. 2019, 25, 1104–1109. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Moretti, C.; Benjamin, N.; Weitzberg, E. Symbiotic bacteria enhance exercise performance. Br. J. Sports Med. 2021, 55, 243. [Google Scholar] [CrossRef]
- Wen, X.; Luo, S.; Lv, D.; Jia, C.; Zhou, X.; Zhai, Q.; Xi, L.; Yang, C. Variations in the fecal microbiota and their functions of Thoroughbred, Mongolian, and Hybrid horses. Front. Veter- Sci. 2022, 9, 920080. [Google Scholar] [CrossRef]
- Bermon, S.; Petriz, B.; Kajėnienė, A.; Prestes, J.; Castell, L.; Franco, O.L. The microbiota: An exercise immunology perspective. Exerc. Immunol. Rev. 2015, 21, 70–79. [Google Scholar] [PubMed]
- Evans, C.C.; LePard, K.J.; Kwak, J.W.; Stancukas, M.C.; Laskowski, S.; Dougherty, J.; Moulton, L.; Glawe, A.; Wang, Y.; Leone, V.; et al. Exercise Prevents Weight Gain and Alters the Gut Microbiota in a Mouse Model of High Fat Diet-Induced Obesity. PLoS ONE 2014, 9, e92193. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Keohane, D.M.; Woods, T.; O’connor, P.; Underwood, S.; Cronin, O.; Whiston, R.; O’Sullivan, O.; Cotter, P.; Shanahan, F.; Molloy, M.G. Four men in a boat: Ultra-endurance exercise alters the gut microbiome. J. Sci. Med. Sport 2019, 22, 1059–1064. [Google Scholar] [CrossRef]
- Janabi, A.; Biddle, A.; Klein, D.; McKeever, K. Exercise training-induced changes in the gut microbiota of Standardbred racehorses. Comp. Exerc. Physiol. 2016, 12, 119–130. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, B.; Bai, D.; Huang, J.; Shiraigo, W.; Yang, L.; Zhao, Q.; Ren, X.; Wu, J.; Bao, W.; et al. Comparison of fecal microbiota of Mongolian and Thoroughbred Horses by high-throughput sequencing of the V4 Region of the 16S rRNA gene. Asian-Australasian J. Anim. Sci. 2015, 29, 1345–1352. [Google Scholar] [CrossRef]
- Petersen, L.M.; Bautista, E.J.; Nguyen, H.; Hanson, B.M.; Chen, L.; Lek, S.H.; Sodergren, E.; Weinstock, G.M. Community characteristics of the gut microbiomes of competitive cyclists. Microbiome 2017, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Park, Y.M.; Holscher, H.D.; Padilla, J.; Scroggins, R.J.; Welly, R.; Britton, S.L.; Koch, L.G.; Vieira-Potter, V.J.; Swanson, K.S. Physical activity differentially affects the cecal microbiota of ovariectomized female rats selectively bred for high and low aerobic capacity. PLoS ONE 2015, 10, e0136150. [Google Scholar] [CrossRef]
- Morita, E.; Yokoyama, H.; Imai, D.; Takeda, R.; Ota, A.; Kawai, E.; Hisada, T.; Emoto, M.; Suzuki, Y.; Okazaki, K. Aerobic exercise training with brisk walking increases intestinal bacteroides in healthy elderly women. Nutrients 2019, 11, 868. [Google Scholar] [CrossRef]
- Denou, E.; Marcinko, K.; Surette, M.G.; Steinberg, G.R.; Schertzer, J.D. High-intensity exercise training increases the diversity and metabolic capacity of the mouse distal gut microbiota during diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E982–E993. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Ruet, A.; Clark, A.; Bars-Cortina, D.; Ramayo-Caldas, Y.; Crisci, E.; Pennarun, S.; Dhorne-Pollet, S.; Foury, A.; Moisan, M.-P.; et al. Priming for welfare: Gut microbiota is associated with equitation conditions and behavior in horse athletes. Sci. Rep. 2020, 10, 8311. [Google Scholar] [CrossRef]
- Plancade, S.; Clark, A.; Philippe, C.; Helbling, J.-C.; Moisan, M.-P.; Esquerré, D.; Le Moyec, L.; Robert, C.; Barrey, E.; Mach, N. Unraveling the effects of the gut microbiota composition and function on horse endurance physiology. Sci. Rep. 2019, 9, 9620. [Google Scholar] [CrossRef]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Trained Group | Untrained Group |
|---|---|---|
| Results of the 30th test event (s) | 302.33 ± 21.30 B | 366.04 ± 25.46 A |
| Results of the 60th test event (s) | 290.64 ± 15.59 B | 357.93 ± 10.22 A |
| Sample Name | Raw Reads | Clean Reads | Q20/% | Q30/% | GC/% | Effective Rate/% |
|---|---|---|---|---|---|---|
| Q.72Y | 6653.59 | 6554.32 | 98.47 | 94.97 | 47.27 | 98.508 |
| Q.80Y | 6729.13 | 6618.38 | 98.21 | 94.2 | 43.64 | 98.354 |
| Q.88Y | 6645.57 | 6525.99 | 98.31 | 94.55 | 47.8 | 98.201 |
| Q.93Y | 6492.36 | 6393.68 | 98.42 | 94.81 | 45.63 | 98.48 |
| Q.104Y | 6838.43 | 6733.09 | 98.31 | 94.45 | 42.56 | 98.46 |
| Q.105Y | 6929.09 | 6783.72 | 98.05 | 93.83 | 38.81 | 97.902 |
| Q.73B | 5963.66 | 5856.69 | 98.22 | 94.28 | 45.98 | 98.206 |
| Q.86B | 5859.63 | 5768.79 | 98.41 | 94.78 | 45.19 | 98.45 |
| Q.91B | 5925.94 | 5809.46 | 98.08 | 93.88 | 46.05 | 98.034 |
| Q.97B | 6220.65 | 6125.11 | 98.35 | 94.61 | 46.4 | 98.464 |
| H.72Y | 6696.04 | 6577.68 | 98.22 | 94.26 | 45.86 | 98.232 |
| H.80Y | 6825.94 | 6731.50 | 98.41 | 94.73 | 42.8 | 98.616 |
| H.88Y | 6839.76 | 6737.69 | 98.42 | 94.79 | 44.2 | 98.508 |
| H.93Y | 6459.27 | 6365.68 | 98.41 | 94.78 | 43.8 | 98.551 |
| H.104Y | 6294.44 | 6195.70 | 98.4 | 94.75 | 44.31 | 98.431 |
| H.105Y | 6178.19 | 6054.10 | 98.19 | 94.18 | 43.05 | 97.991 |
| H.73B | 6595.90 | 6470.27 | 98.35 | 94.62 | 43.59 | 98.095 |
| H.86B | 6634.70 | 6540.49 | 98.39 | 94.69 | 43.72 | 98.58 |
| H.91B | 6458.46 | 6351.98 | 98.23 | 94.27 | 43.47 | 98.351 |
| H.97B | 6687.72 | 6567.63 | 98.31 | 94.54 | 46.37 | 98.204 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-F.; Han, Z.-X.; Yao, X.-K.; Meng, J.; Ren, W.-L.; Wang, C.-K.; Yuan, X.-X.; Zeng, Y.-Q.; Wang, Y.-F.; Sun, Z.-W.; et al. Effects of Different Stages of Training on the Intestinal Microbes of Yili Horses Analyzed Using Metagenomics. Genes 2025, 16, 504. https://doi.org/10.3390/genes16050504
Sun Y-F, Han Z-X, Yao X-K, Meng J, Ren W-L, Wang C-K, Yuan X-X, Zeng Y-Q, Wang Y-F, Sun Z-W, et al. Effects of Different Stages of Training on the Intestinal Microbes of Yili Horses Analyzed Using Metagenomics. Genes. 2025; 16(5):504. https://doi.org/10.3390/genes16050504
Chicago/Turabian StyleSun, Yuan-Fang, Zi-Xiang Han, Xin-Kui Yao, Jun Meng, Wan-Lu Ren, Chuan-Kun Wang, Xin-Xin Yuan, Ya-Qi Zeng, Yong-Fa Wang, Zhi-Wen Sun, and et al. 2025. "Effects of Different Stages of Training on the Intestinal Microbes of Yili Horses Analyzed Using Metagenomics" Genes 16, no. 5: 504. https://doi.org/10.3390/genes16050504
APA StyleSun, Y.-F., Han, Z.-X., Yao, X.-K., Meng, J., Ren, W.-L., Wang, C.-K., Yuan, X.-X., Zeng, Y.-Q., Wang, Y.-F., Sun, Z.-W., & Wang, J.-W. (2025). Effects of Different Stages of Training on the Intestinal Microbes of Yili Horses Analyzed Using Metagenomics. Genes, 16(5), 504. https://doi.org/10.3390/genes16050504