
Physiological Responses and Transcriptome Analysis of Camellia reticulata Under Low-Temperature Stress

Abstract
1. Introduction
2. Materials and Methods
2.1. Treatment of Test Materials
2.2. Measurement of Physiological Indexes
2.3. Transcriptome Sequencing and Data Analysis
2.4. QRT-PCR Analysis
3. Results
3.1. Effect and Analysis of Cold Stress on Physiology of C. reticulata Slices
3.1.1. Change of Soluble Substance Content in Low Temperature Stress Treatment
3.1.2. Changes of Chlorophyll Content in Low-Temperature Stress Treatment
3.1.3. Changes of Peroxidase Activity During Low-Temperature Stress Treatment
3.1.4. Change of Malondialdehyde Content in Low-Temperature Stress Treatment
3.1.5. Correlation Analysis of Physiological Indicators
3.2. Transcriptome Analysis of C. reticulata ‘Shizhitou’ Leaves Under Cold Stress
3.2.1. Sequencing and Annotation
3.2.2. Analysis of Gene Expression Distribution
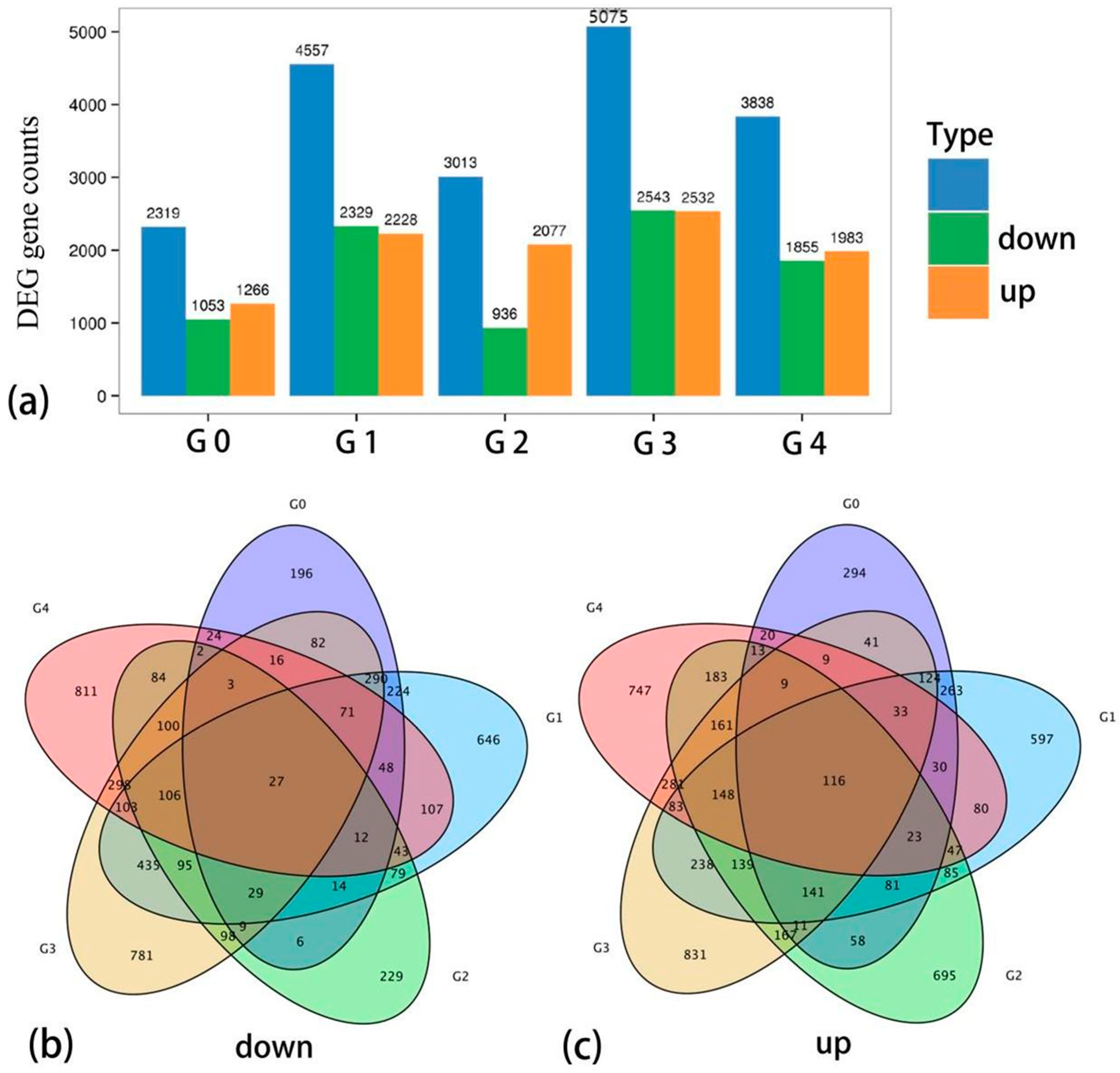
3.2.3. Differential Expression Gene Analysis
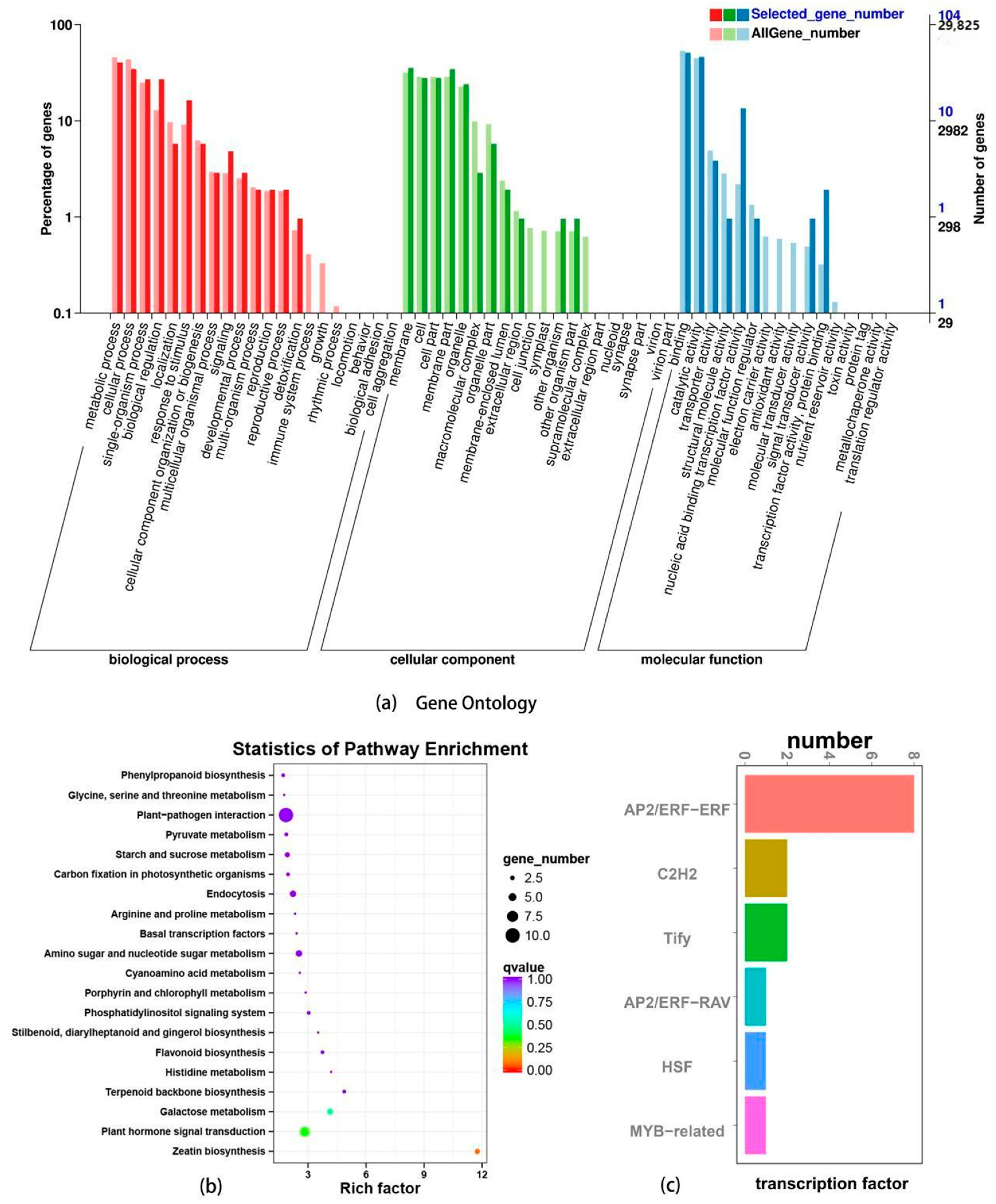
3.2.4. GO Annotation, KEGG Enrichment, and Transcription Factor Analysis of Differentially Expressed Genes
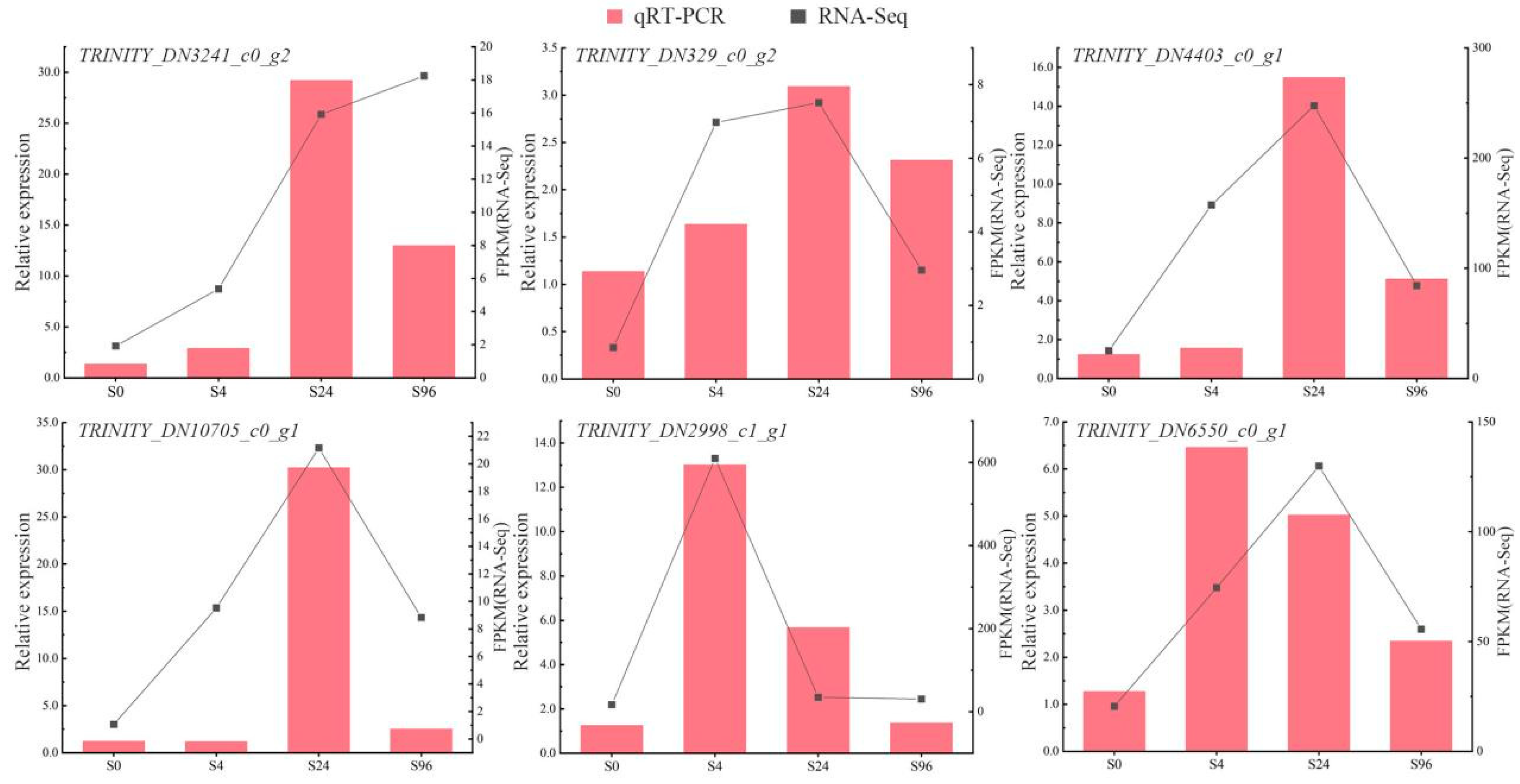
3.2.5. Localization Gene Verification
4. Discussion
4.1. Physiological Responses to Cold Stress in C. reticulata
4.2. Transcriptomic Mechanisms Underlying Cold Stress Responses
4.3. Physiology-Transcriptome Integration: Unifying Mechanisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; Zhang, X.; Li, Y.; Tian, X.; Tian, X.; Zhao, H.; Xuan, Z.; Xue, K.; Li, Y.; Lin, W. Molecular mechanisms underlying floral fragrance in Camellia japonica ‘High Fragrance’: A time-course assessment. Front. Plant Sci. 2024, 15, 1461442. [Google Scholar] [CrossRef]
- Zheng, J.; Su, H.; Pu, S.; Chen, H.; El-Kassaby, Y.A.; Yang, Z.; Feng, J. High-yield hybrid breeding of Camellia oleifolia based on ISSR molecular markers. BMC Plant Biol. 2024, 24, 517. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Y.; Jia, X. The complete chloroplast genome of Camellia osmantha, an edible oil Camellia. Mitochondrial DNA. Part B Resour. 2021, 6, 3169–3170. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; de Riek, J.; Guo, H.; Jarvis, D.; Ma, L.; Long, C. Impact of traditional culture on Camellia reticulata in Yunnan, China. J. Ethnobiol. Ethnomed. 2015, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; Huang, W.; De Riek, J.; Zhang, S.; Ahmed, S.; Van Huylenbroeck, J.; Long, C. Genetic diversity, population structure, and traditional culture of Camellia reticulata. Ecol. Evol. 2017, 7, 8915–8926. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Du, Y.; Zhao, Z.; Zhu, X.; Jiang, X.; Shu, Z.; Yin, Y.; Li, X. Overexpression of Camellia sinensis H1 histone gene confers abiotic stress tolerance in transgenic tobacco. Plant Cell Rep. 2014, 33, 1829–1841. [Google Scholar] [CrossRef]
- Li, M.; Wang, W.; Chen, X.; Lu, X.; Huang, Y. Combining resistance indicators, metabolomes and transcriptomes to reveal correlations in disease and cold resistance in tea plant and analyze the key domain NB-ARC. Plant Cell Rep. 2025, 44, 34. [Google Scholar] [CrossRef]
- Zaman, S.; Shen, J.; Wang, S.; Song, D.; Wang, H.; Ding, S.; Pang, X.; Wang, M.; Sabir, I.A.; Wang, Y.; et al. Effect of shading on physiological attributes and comparative transcriptome analysis of Camellia sinensis cultivar reveals tolerance mechanisms to low temperatures. Front. Plant Sci. 2023, 14, 1114988. [Google Scholar] [CrossRef]
- Wan, S.; Wang, W.; Zhou, T.; Zhang, Y.; Chen, J. Transcriptomic analysis reveals the molecular mechanisms of Camellia sinensis in response to salt stress. Plant Growth Regul. 2018, 84, 481–492. [Google Scholar] [CrossRef]
- Dai, X.; Zhao, G.; Jiao, T.; Wu, Y.; Li, X.; Zhou, K.; Gao, L.; Xia, T. Involvement of Three CsRHM Genes from Camellia sinensis in UDP-Rhamnose Biosynthesis. J. Agric. Food Chem. 2018, 66, 7139–7149. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, C.J.; Li, Y.Y.; Wei, C.L.; Deng, W.W. CsICE1 and CsCBF1: Two transcription factors involved in cold responses in Camellia sinensis. Plant Cell Rep. 2012, 31, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Müller, M.; Bai, T.; Yao, S.; Liu, Z. Transcriptome profiling in Camellia japonica var. decumbens for the discovery of genes involved in chilling tolerance under cold stress. Ann. For. Res. 2019, 62. [Google Scholar] [CrossRef]
- Chain, F.; Côté-Beaulieu, C.; Belzile, F.; Menzies, J.G.; Bélanger, R.R. A comprehensive transcriptomic analysis of the effect of silicon on wheat plants under control and pathogen stress conditions. Mol. Plant-Microbe Interact. MPMI 2009, 22, 1323. [Google Scholar] [CrossRef]
- Chen, H.; Liang, X.; Yang, Z. Effects of Low-Temperature Stress on Physiological Characteristics and Microstructure of Stems and Leaves of Pinus massoniana L. Plants 2024, 13, 2229. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, M.; Zhou, Y.; Wu, Q.; Shen, Y.; Cui, Z.; Liu, R.; Liu, R.; Shen, Q.; Chen, J. Freezing transcriptome analysis showed that GhZAT10 regulates freezing tolerance through a partially CBF-dependent pathway in upland cotton (Gossypium hirsutum L.). Environ. Exp. Bot. 2023, 208, 105263. [Google Scholar] [CrossRef]
- Zhang, C.W.; Wei, Y.P.; Xiao, D.; Gao, L.W.; Lyu, S.W.; Hou, X.L.; Bouuema, G. Transcriptomic and proteomic analyses provide new insights into the regulation mechanism of low-temperature-induced leafy head formation in Chinese cabbage. J. Proteom. 2016, 144, 1–10. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, S.; Wang, F.; Lu, J.; Tan, G.; Wang, N.; Qi, F.; Zhang, C.; Deyholos, M.K.; Zang, Z. Transcriptome analysis and physiological response to heat and cold stress in flax (Linum usitatissimum L) at the seedling stage. Environ. Exp. Bot. 2025, 229, 106076. [Google Scholar] [CrossRef]
- Zhao, D.; Jiang, Y.; Ning, C.; Meng, J.; Lin, S.; Ding, W. Transcriptome sequencing of a chimaera reveals coordinated expression of anthocyanin biosynthetic genes mediating yellow formation in herbaceous peony (Paeonia lactiflora Pall.). Bmc Genom. 2014, 15, 689. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Yangyang, D.; Jianqi, L.I.; Songfeng, W.U.; Yunping, Z.; Yaowen, C.; Fuchu, H.E. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Dewey, C.N.; Bo, L. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Wang, W.; Gao, T.; Chen, J.; Yang, J.; Huang, H.; Yu, Y. The late embryogenesis abundant gene family in tea plant (Camellia sinensis): Genome-wide characterization and expression analysis in response to cold and dehydration stress. Plant Physiol. Biochem. 2019, 135, 277–286. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Li, T.; Teng, R.M.; Han, M.H.; Zhuang, J. Low temperature effects on carotenoids biosynthesis in the leaves of green and albino tea plant (Camellia sinensis (L.) O. Kuntze). Sci. Hortic. 2021, 285, 110164. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.W.; Wu, Z.J.; Wang, Y.X.; Teng, R.M.; Zhuang, J. Differentially expressed protein and gene analysis revealed the effects of temperature on changes in ascorbic acid metabolism in harvested tea leaves. Hortic. Res. 2018, 5, 65. [Google Scholar] [CrossRef]
- Wu, L.; Li, J.; Li, Z.; Zhang, F.; Tan, X. Transcriptomic Analyses of Camellia oleifera ’Huaxin’ Leaf Reveal Candidate Genes Related to Long-Term Cold Stress. Int. J. Mol. Sci. 2020, 21, 846. [Google Scholar] [CrossRef]
- Wang, X.C.; Zhao, Q.Y.; Ma, C.L.; Zhang, Z.H.; Cao, H.L.; Kong, Y.M.; Yue, C.; Hao, X.Y.; Chen, L.; Ma, J.Q.; et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genom. 2013, 14, 415. [Google Scholar] [CrossRef]
- Luo, J.; Huang, S.; Chang, Y.; Li, H.; Guo, G. Physiological and transcriptomic analyses reveal tea plant (Camellia sinensis L.) adapts to extreme freezing stress during winter by regulating cell wall structure. BMC Genom. 2023, 24, 558. [Google Scholar] [CrossRef]
- Fan, M.; Yang, K.; Zhou, R.; Liu, Q.; Guo, X.; Sun, Y. Temporal transcriptome profiling reveals candidate genes involved in cold acclimation of Camellia japonica (Naidong). Plant Physiol. Biochem. 2021, 167, 795–805. [Google Scholar] [CrossRef]
- Kidokoro, S.; Kim, J.S.; Ishikawa, T.; Suzuki, T.; Shinozaki, K.; Yamaguchi-Shinozaki, K. DREB1A/CBF3 Is Repressed by Transgene-Induced DNA Methylation in the Arabidopsis ice1-1 Mutant. Plant Cell 2020, 32, 1035–1048. [Google Scholar] [CrossRef]
- Liu, Z.; Bi, S.; Meng, J.; Liu, T.; Li, P.; Yu, C.; Peng, X. Arbuscular mycorrhizal fungi enhanced rice proline metabolism under low temperature with nitric oxide involvement. Front. Plant Sci. 2022, 13, 962460. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Ban, Q.; Zhu, X.; Jiang, C.; Wei, C.; Bennetzen, J.L. Comparative transcriptomic analysis reveals gene expression associated with cold adaptation in the tea plant Camellia sinensis. BMC Genom. 2019, 20, 624. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.; Vigmond, E.J.; Plane, F.; Welsh, D.G. Mechanistic basis of differential conduction in skeletal muscle arteries. J. Physiol. 2009, 587, 1301–1318. [Google Scholar] [CrossRef] [PubMed]
- Cortleven, A.; Leuendorf, J.E.; Frank, M.; Pezzetta, D.; Bolt, S.; Schmülling, T. Cytokinin action in response to abiotic and biotic stresses in plants. Plant Cell Environ. 2019, 42, 998–1018. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hwarari, D.; Korboe, H.M.; Ahmad, B.; Cao, Y.; Movahedi, A.; Yang, L. Low temperature stress-induced perception and molecular signaling pathways in plants. Environ. Exp. Bot. 2023, 207, 105190. [Google Scholar] [CrossRef]
- Li, Q.; Lei, S.; Du, K.; Li, L.; Pang, X.; Wang, Z.; Wei, M.; Fu, S.; Hu, L.; Xu, L. RNA-seq based transcriptomic analysis uncovers α-linolenic acid and jasmonic acid biosynthesis pathways respond to cold acclimation in Camellia japonica. Sci. Rep. 2016, 6, 36463. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Primer Sequence (5′→3′) |
|---|---|---|
| HSF | TRINITY_DN3241_c0_g2 | F: CGCTTGAAGCTTTACAGGGC |
| R: TTCCTCGGAAGTACGAGCCT | ||
| CCT | TRINITY_DN329_c0_g2 | F: CGGACATCCATAAGGCGACA |
| R: CGTTTTCGATGTTCCGAGGC | ||
| TIFY | TRINITY_DN4403_c0_g1 | F: AACTCCGGTGACGAGAAACC |
| R: CTTTGCCTTATCAGCCGGGA | ||
| AP2 | TRINITY_DN10705_c0_g1 | F: AGGGGTCGTAGGCTATGGTT |
| R: TGGCTCGAAGTTGTTGTGGA | ||
| C2H2 | TRINITY_DN2998_c1_g1 | F:GGCAAACTCTAGCCCGCATA |
| R: GAGACCACATCGACCAAGGG | ||
| AP2 | TRINITY_DN6550_c0_g1 | F: ACGTCTTTTCCGGCGATTCT |
| R: GAGGATTTGGCTCGGGACTT |
| Soluble Protein | Soluble Sugar | MDA | Chlorophyll a | Chlorophyll b | Total Chlorophyll | Proline | POD | CAT | |
|---|---|---|---|---|---|---|---|---|---|
| Soluble protein | 1 | ||||||||
| Soluble sugar | 0.493 * | 1 | |||||||
| MDA | −0.131 | −0.108 | 1 | ||||||
| chlorophyll a | −0.269 | 0.041 | 0.096 | 1 | |||||
| chlorophyll b | −0.473 * | −0.146 | 0.07 | 0.963 ** | 1 | ||||
| total chlorophyll | −0.336 | −0.018 | 0.088 | 0.996 ** | 0.983 ** | 1 | |||
| proline | −0.445 | −0.373 | 0.132 | −0.193 | −0.079 | −0.158 | 1 | ||
| POD | −0.072 | −0.005 | −0.118 | −0.512 * | −0.486 * | −0.508 * | 0.283 | 1 | |
| CAT | 0.404 | 0.215 | −0.068 | −0.199 | −0.24 | −0.214 | −0.291 | −0.555 * | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Dong, J.; Pu, R.; Wang, P.; Sun, T.; Li, J.; Zhang, J.; Bai, T. Physiological Responses and Transcriptome Analysis of Camellia reticulata Under Low-Temperature Stress. Genes 2025, 16, 503. https://doi.org/10.3390/genes16050503
Wu Y, Dong J, Pu R, Wang P, Sun T, Li J, Zhang J, Bai T. Physiological Responses and Transcriptome Analysis of Camellia reticulata Under Low-Temperature Stress. Genes. 2025; 16(5):503. https://doi.org/10.3390/genes16050503
Chicago/Turabian StyleWu, Yawen, Jian Dong, Ran Pu, Pan Wang, Timei Sun, Jie Li, Jingli Zhang, and Tian Bai. 2025. "Physiological Responses and Transcriptome Analysis of Camellia reticulata Under Low-Temperature Stress" Genes 16, no. 5: 503. https://doi.org/10.3390/genes16050503
APA StyleWu, Y., Dong, J., Pu, R., Wang, P., Sun, T., Li, J., Zhang, J., & Bai, T. (2025). Physiological Responses and Transcriptome Analysis of Camellia reticulata Under Low-Temperature Stress. Genes, 16(5), 503. https://doi.org/10.3390/genes16050503