Mouse ENU Mutagenesis to Understand Immunity to Infection: Methods, Selected Examples, and Perspectives

Abstract
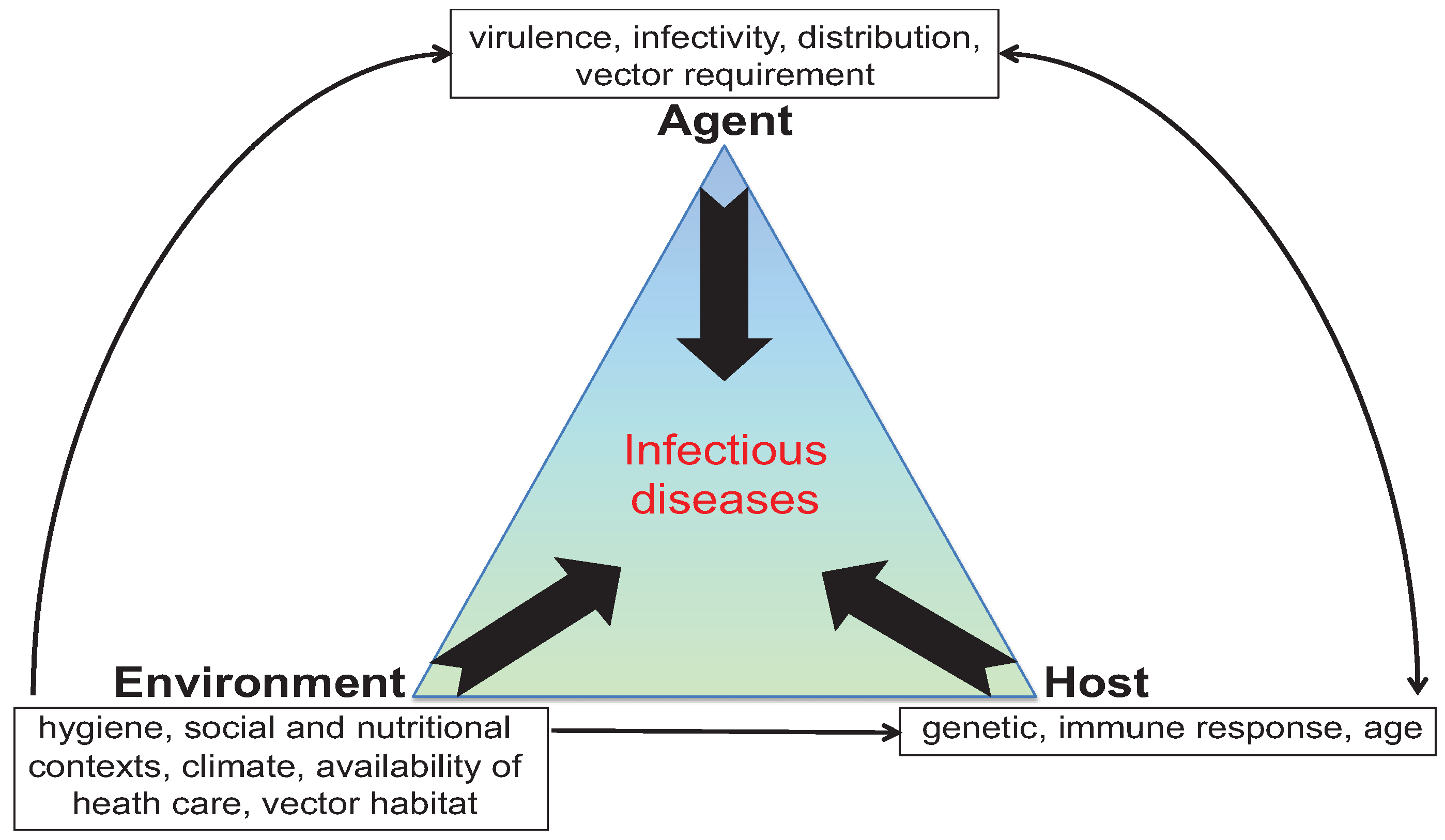
:1. Introduction

2. Mice to the Rescue
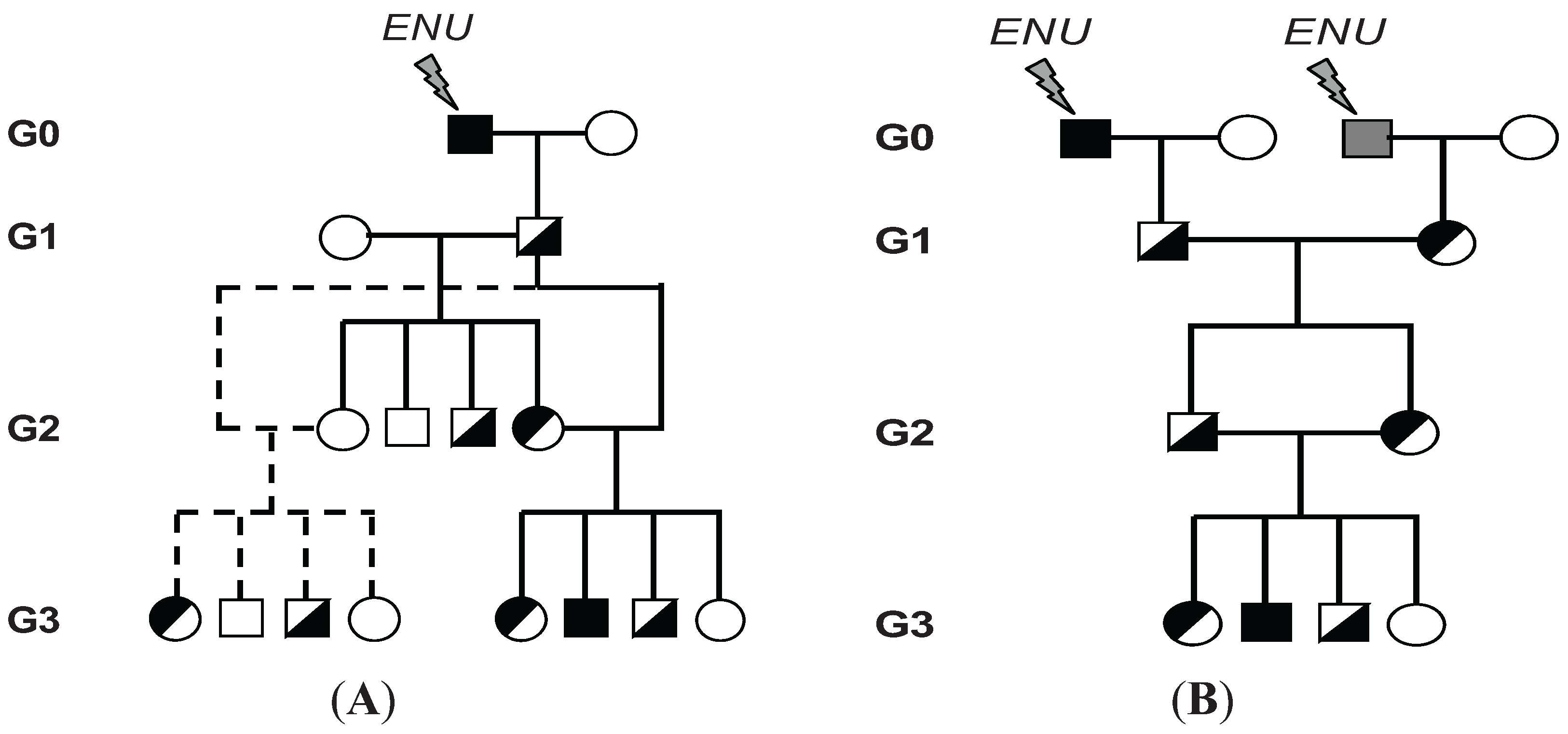
3. Chemical Mutagenesis and Generation of Mice Carrying Homozygous ENU-Induced Mutations

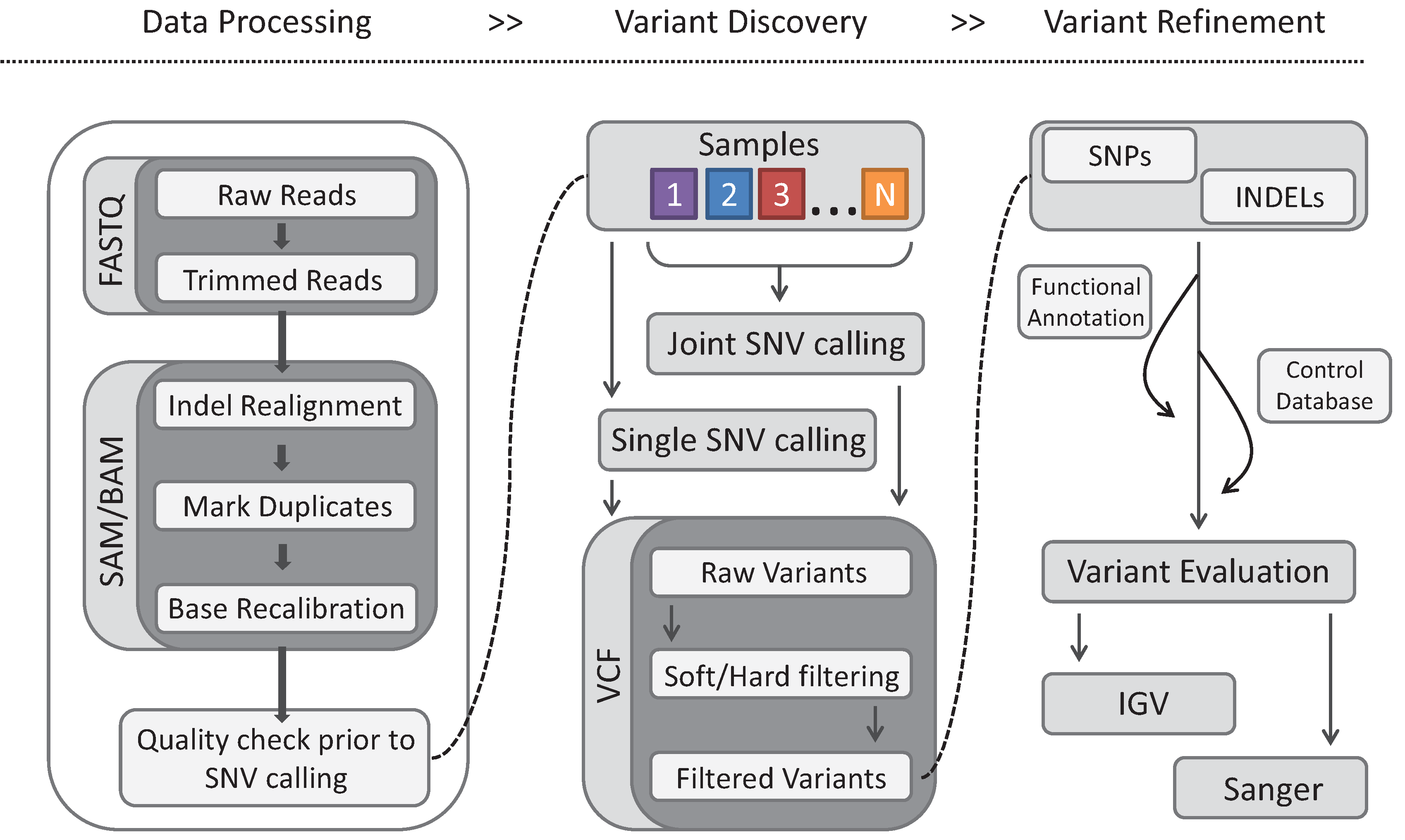
4. Gene Identification

{kind=link}
{kind=link}
{kind=link}
| Agilent Sureselect Mouse All Exon | Nimblegen SeqCap Ez | |
|---|---|---|
| Probe size | 120 bases | 55–105 bases |
| Target Region size | 49.6 Mb | 54.3 Mb |
| Probe Type | RNA | DNA |
| Number of Targeted Exons | 221,784 | 203,225 |

5. Infectious Screens
6. Malaria Parasites
6.1. Screening for Acquired Resistance to Cerebral Malaria
6.2. Screening for Acquired Resistance to Blood-Stage Malaria
6.3. Conclusion
7. Salmonella Bacteria Infections
7.1. Screening for Acquired Susceptibility to Salmonella typhimurium
| Malaria | Salmonella | HSV-1 | |
|---|---|---|---|
| G1 males | 573 | 491 | 265 |
| G3 mice | 16,411 | 8,415 | 7,802 |
| Deviant pedigrees (in progress) | 45 | 16 | 11 |
| Confirmed pedigrees | 5 | 3 | 2 |
7.2. Ex Vivo and in Vivo ENU Screens for Susceptibility to Bacteria Infections
7.3. Conclusion
8. Herpes Viruses
8.1. Cytomegaloviruses
8.2. Screening for Altered Immune Responses to MCMV
8.3. Herpes Simplex Virus 1
8.4. Screening for Acquired Susceptibility to HSE
9. Conclusions and Perspectives
| Pathway | Gene | Screen | Phenotype | Reference |
|---|---|---|---|---|
| TLR signaling | Cd36 | Immunity→S. aureus | Susceptible | [165] |
| Map3k8 | Immunity→Group B streptococcus | Susceptible | [166] | |
| Ptpn6 | Autoimmunity→L. monocytogenes | Susceptible | [172] | |
| Tlr9 | Immunity→MCMV | Susceptible | [179] | |
| Trif | Immunity→MCMV | Susceptible | [180] | |
| Unc93b1 | Immunity→MCMV | Susceptible | [181] | |
| Ikbkg | Immunity→MCMV | Susceptible | [183] | |
| Type I IFN signal | Usp18 | S . Typhimurium | Susceptible | [156,159] |
| Stat1 | MCMV | Susceptible | [182] | |
| Effector | Eif2ak4 | MCMV | Susceptible | [184] |
| Cellular immunity | Jak3 | P. Berghei | Resistant | [119] |
| Foxn1 | P. Berghei | Resistant | [125] | |
| Stat4 | S . Typhimurium | Susceptible | [155,156] | |
| Tnf | Immunity→L.monocytogenes | Resistant | [167] | |
| Gfi1 | Immunity→S. Typhimurium | Susceptible | [170] | |
| Gimap5 | Immunity→MCMV | Susceptible | [194] | |
| Unc13d | MCMV | Susceptible | [195] | |
| Flt3 | MCMV | Susceptible | [197] | |
| Slfn2 | MCMV | Susceptible | [169] | |
| Ptprc | HSV-1 | Susceptible | [209] | |
| Red cell cytoskeleton | Ank1 | S. Typhimurium | Susceptible | [154] |
| Ank1 | P. Chabaudi | Resistant | [127,128] | |
| Homeostasis | Kcnj8 | MCMV | Susceptible | [198] |
| Lipid metabolism | Scd1 | Immunity→S. Pyogenes | Susceptible | [171] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brussow, H. Europe, the bull and the Minotaur: The biological legacy of a Neolithic love story. Environ. Microbiol. 2009, 11, 2778–2788. [Google Scholar]
- McMichael, A.J. Environmental and social influences on emerging infectious diseases: Past, present and future. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004, 359, 1049–1058. [Google Scholar]
- Casanova, J.L.; Abel, L. Inborn errors of immunity to infection: The rule rather than the exception. J. Exp. Med. 2005, 202, 197–201. [Google Scholar]
- Fauci, A.S.; Morens, D.M. The perpetual challenge of infectious diseases. N. Engl. J. Med. 2012, 366, 454–461. [Google Scholar]
- Chapman, S.J.; Hill, A.V. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar]
- Cobat, A.; Orlova, M.; Barrera, L.F.; Schurr, E. Host genomics and control of tuberculosis infection. Publ. Health Genet. 2013, 16, 44–49. [Google Scholar]
- Plantinga, T.S.; Johnson, M.D.; Scott, W.K.; Joosten, L.A.; van der Meer, J.W.; Perfect, J.R.; Kullberg, B.J.; Netea, M.G. Human genetic susceptibility to Candida infections. Med. Mycol. 2012, 50, 785–794. [Google Scholar]
- Keynan, Y.; Malik, S.; Fowke, K.R. The role of polymorphisms in host immune genes in determining the severity of respiratory illness caused by pandemic H1N1 influenza. Publ. Health Genet. 2013, 16, 9–16. [Google Scholar]
- Min-Oo, G.; Gros, P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. 2005, 7, 753–763. [Google Scholar]
- Lederman, M.M.; Penn-Nicholson, A.; Cho, M.; Mosier, D. Biology of CCR5 and its role in HIV infection and treatment. JAMA 2006, 296, 815–826. [Google Scholar]
- Lindesmith, L.; Moe, C.; Marionneau, S.; Ruvoen, N.; Jiang, X.; Lindblad, L.; Stewart, P.; LePendu, J.; Baric, R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003, 9, 548–553. [Google Scholar]
- Von Bernuth, H.; Picard, C.; Puel, A.; Casanova, J.L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 2012, 42, 3126–3135. [Google Scholar]
- International Human Genome Sequencing. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar]
- Hohl, T.M. Overview of vertebrate animal models of fungal infection. J. Immunol. Methods 2014. [Google Scholar] [CrossRef]
- Longley, R.; Smith, C.; Fortin, A.; Berghout, J.; McMorran, B.; Burgio, G.; Foote, S.; Gros, P. Host resistance to malaria: Using mouse models to explore the host response. Mamm. Genet. 2011, 22, 32–42. [Google Scholar]
- Sancho-Shimizu, V.; Zhang, S.Y.; Abel, L.; Tardieu, M.; Rozenberg, F.; Jouanguy, E.; Casanova, J.L. Genetic susceptibility to herpes simplex virus 1 encephalitis in mice and humans. Curr. Opin. Allergy Clin. Immunol. 2007, 7, 495–505. [Google Scholar]
- Wick, M.J. Innate immune control of Salmonella enterica serovar Typhimurium: Mechanisms contributing to combating systemic Salmonella infection. J. Innate Immun. 2011, 3, 543–549. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Church, D.M.; Goodstadt, L.; Hillier, L.W.; Zody, M.C.; Goldstein, S.; She, X.; Bult, C.J.; Agarwala, R.; Cherry, J.L.; DiCuccio, M.; et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009, 7, e1000112. [Google Scholar]
- Guenet, J.L. Animal models of human genetic diseases: Do they need to be faithful to be useful? Mol. Genet. Genet. 2011, 286, 1–20. [Google Scholar]
- Guan, C.; Ye, C.; Yang, X.; Gao, J. A review of current large-scale mouse knockout efforts. Genesis 2010, 48, 73–85. [Google Scholar]
- Ayadi, A.; Birling, M.C.; Bottomley, J.; Bussell, J.; Fuchs, H.; Fray, M.; Gailus-Durner, V.; Greenaway, S.; Houghton, R.; Karp, N.; et al. Mouse large-scale phenotyping initiatives: Overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm. Genome 2012, 23, 600–610. [Google Scholar]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar]
- Blake, J.A.; Bult, C.J.; Eppig, J.T.; Kadin, J.A.; Richardson, J.E. The Mouse Genome Database: Integration of and access to knowledge about the laboratory mouse. Nucl. Acids Res. 2014, 42, D810–D817. [Google Scholar]
- Smith, C.M.; Finger, J.H.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Berghout, J.; Campbell, J.; Corbani, L.E.; Forthofer, K.L.; Frost, J.P. The mouse Gene Expression Database (GXD): 2014 update. Nucl. Acids Res. 2014, 42, D818–D824. [Google Scholar]
- Begley, D.A.; Krupke, D.M.; Neuhauser, S.B.; Richardson, J.E.; Bult, C.J.; Eppig, J.T.; Sundberg, J.P. The Mouse Tumor Biology Database (MTB): A central electronic resource for locating and integrating mouse tumor pathology data. Vet. Pathol. 2012, 49, 218–223. [Google Scholar]
- Wiltshire, S.A.; Leiva-Torres, G.A.; Vidal, S.M. Quantitative trait locus analysis, pathway analysis, and consomic mapping show genetic variants of Tnni3k, Fpgt, or H28 control susceptibility to viral myocarditis. J. Immunol. 2011, 186, 6398–6405. [Google Scholar]
- Di Pietrantonio, T.; Hernandez, C.; Girard, M.; Verville, A.; Orlova, M.; Belley, A.; Behr, M.A.; Loredo-Osti, J.C.; Schurr, E. Strain-specific differences in the genetic control of two closely related mycobacteria. PLoS Pathog. 2010, 6, e1001169. [Google Scholar]
- Toth, L.A.; Trammell, R.A.; Williams, R.W. Mapping complex traits using families of recombinant inbred strains: An overview and example of mapping susceptibility to Candida albicans induced illness phenotypes. Pathog. Dis. 2014, 71, 232–246. [Google Scholar]
- Boivin, G.A.; Pothlichet, J.; Skamene, E.; Brown, E.G.; Loredo-Osti, J.C.; Sladek, R.; Vidal, S.M. Mapping of clinical and expression quantitative trait loci in a sex-dependent effect of host susceptibility to mouse-adapted influenza H3N2/HK/1/68. J. Immunol. 2012, 188, 3949–3960. [Google Scholar]
- Ferris, M.T.; Aylor, D.L.; Bottomly, D.; Whitmore, A.C.; Aicher, L.D.; Bell, T.A.; Bradel-Tretheway, B.; Bryan, J.T.; Buus, R.J.; Gralinski, L.E.; et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013, 9, e1003196. [Google Scholar]
- Guenet, J.L.; Bonhomme, F. Wild mice: An ever-increasing contribution to a popular mammalian model. Trends Genet. 2003, 19, 24–31. [Google Scholar]
- Chia, R.; Achilli, F.; Festing, M.F.; Fisher, E.M. The origins and uses of mouse outbred stocks. Nat. Genet. 2005, 37, 1181–1186. [Google Scholar]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011, 477, 289–294. [Google Scholar]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genet. Biol. 2013, 14, R82. [Google Scholar]
- Vidal, S.M.; Malo, D.; Marquis, J.F.; Gros, P. Forward genetic dissection of immunity to infection in the mouse. Annu. Rev. Immunol. 2008, 26, 81–132. [Google Scholar]
- Cook, M.C.; Vinuesa, C.G.; Goodnow, C.C. ENU-mutagenesis: Insight into immune function and pathology. Curr. Opin. Immunol. 2006, 18, 627–633. [Google Scholar]
- Hoebe, K.; Beutler, B. Forward genetic analysis of TLR-signaling pathways: An evaluation. Adv. Drug Deliv. Rev. 2008, 60, 824–829. [Google Scholar]
- Hoyne, G.F.; Goodnow, C.C. The use of genomewide ENU mutagenesis screens to unravel complex mammalian traits: Identifying genes that regulate organ-specific and systemic autoimmunity. Immunol. Rev. 2006, 210, 27–39. [Google Scholar]
- Oliver, P.L.; Davies, K.E. New insights into behaviour using mouse ENU mutagenesis. Hum. Mol. Genet. 2012, 21, R72–R81. [Google Scholar]
- Russell, W.L. Radiation and chemical mutagenesis and repair in mice. Johns Hopkins Med. J. Suppl. 1972, 1, 239–247. [Google Scholar]
- Jaenisch, R. Germ line integration and Mendelian transmission of the exogenous Moloney leukemia virus. Proc. Natl. Acad. Sci. USA 1976, 73, 1260–1264. [Google Scholar]
- Russell, L.B.; Russell, W.L. Frequency and nature of specific-locus mutations induced in female mice by radiations and chemicals: A review. Mutat. Res. 1992, 296, 107–127. [Google Scholar]
- Russell, W.L.; Kelly, E.M.; Hunsicker, P.R.; Bangham, J.W.; Maddux, S.C.; Phipps, E.L. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 5818–5819. [Google Scholar]
- Russell, W.L.; Hunsicker, P.R.; Carpenter, D.A.; Cornett, C.V.; Guinn, G.M. Effect of dose fractionation on the ethylnitrosourea induction of specific-locus mutations in mouse spermatogonia. Proc. Natl. Acad. Sci. USA 1982, 79, 3592–3593. [Google Scholar]
- Hitotsumachi, S.; Carpenter, D.A.; Russell, W.L. Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc. Natl. Acad. Sci. USA 1985, 82, 6619–6621. [Google Scholar]
- Singer, B. All oxygens in nucleic acids react with carcinogenic ethylating agents. Nature 1976, 264, 333–339. [Google Scholar]
- Bignami, M.; Vitelli, A.; di Muccio, A.; Terlizzese, M.; Calcagnile, A.; Zapponi, G.A.; Lohman, P.H.M.; den Engelse, L.; Dogliotti1, E. Relationship between specific alkylated bases and mutations at two gene loci induced by ethylnitrosourea and diethyl sulfate in CHO cells. Mutat. Res. 1988, 193, 43–51. [Google Scholar]
- Van Zeeland, A.A. Molecular dosimetry of alkylating agents: Quantitative comparison of genetic effects on the basis of DNA adduct formation. Mutagenesis 1988, 3, 179–191. [Google Scholar]
- Vogel, E.W.; Natarajan, A.T. DNA damage and repair in somatic and germ cells in vivo. Mutat. Res. 1995, 330, 183–208. [Google Scholar]
- Fairfield, H.; Gilbert, G.J.; Barter, M.; Corrigan, R.R.; Curtain, M.; Ding, Y.M.; Ascenzo, M.D.; Gerhardt, D.J.; He, C.; Huang, W.H.; et al. Mutation discovery in mice by whole exome sequencing. Genet. Biol. 2011, 12, R86. [Google Scholar]
- Andrews, T.D.; Whittle, B.; Field, M.A.; Balakishnan, B.; Zhang, Y.; Shao, Y.; Cho, V.; Kirk, M.; Singh, M.; Xia, Y.; et al. Massively parallel sequencing of the mouse exome to accurately identify rare, induced mutations: An immediate source for thousands of new mouse models. Open Biol. 2012, 2, 120061. [Google Scholar]
- Bull, K.R.; Rimmer, A.J.; Siggs, O.M.; Miosge, L.A.; Roots, C.M.; Enders, A.; Bertram, E.M.; Crockford, T.L.; Whittle, B.; Potter, P.K.; et al. Unlocking the bottleneck in forward genetics using whole-genome sequencing and identity by descent to isolate causative mutations. PLoS Genet. 2013, 9, e1003219. [Google Scholar]
- Lewis, M.A.; Quint, E.; Glazier, A.M.; Fuchs, H.; de Angelis, M.H.; Langford, C.; van Dongen, S.; Abreu-Goodger, C.; Piipari, M.; Redshaw, N.; et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat. Genet. 2009, 41, 614–618. [Google Scholar]
- Masuya, H.; Sezutsu, H.; Sakuraba, Y.; Sagai, T.; Hosoya, M.; Kanedaa, H.; Miuraa, I.; Kobayashia, K.; Sumiyamad, K.; Shimizu, A.; et al. A series of ENU-induced single-base substitutions in a long-range cis-element altering sonic hedgehog expression in the developing mouse limb bud. Genomics 2007, 89, 207–214. [Google Scholar]
- Arnold, C.N.; Barnes, M.J.; Berger, M.; Blasius, A.L.; Brandl, K.; Croker, B.; Crozat, K.; Du, X.; Eidenschenk, C.; Georgel, P.; et al. ENU-induced phenovariance in mice: Inferences from 587 mutations. BMC Res. Notes 2012, 5, 577. [Google Scholar]
- Puk, O.; Moller, G.; Geerlof, A.; Krowiorz, K.; Ahmad, N.; Wagner, S.; Adamski, J.; de Angelis, M.H.; Graw, J. The pathologic effect of a novel neomorphic Fgf9(Y162C) allele is restricted to decreased vision and retarded lens growth. PLoS ONE 2011, 6, e23678. [Google Scholar]
- Caspary, T. Phenotype-driven mouse ENU mutagenesis screens. Methods Enzymol. 2010, 477, 313–327. [Google Scholar]
- Probst, F.J.; Justice, M.J. Mouse mutagenesis with the chemical supermutagen ENU. Methods Enzymol. 2010, 477, 297–312. [Google Scholar]
- Georgel, P.; Du, X.; Hoebe, K.; Beutler, B. ENU mutagenesis in mice. Methods Mol. Biol. 2008, 415, 1–16. [Google Scholar]
- Justice, M.J.; Carpenter, D.A.; Favor, J.; Neuhauser-Klaus, A.; de Angelis, M.H.; Soewarto, D.; Moser, A.; Cordes, S.; Miller, D.; Chapman, V.; et al. Effects of ENU dosage on mouse strains. Mamm. Genet. 2000, 11, 484–488. [Google Scholar]
- Bainbridge, M.N.; Wang, M.; Burgess, D.L.; Kovar, C.; Rodesch, M.J.; D’Ascenzo, M.; Kitzman, J.; Wu, Y.-Q.; Newsham, I.; Richmond, T.A.; et al. Whole exome capture in solution with 3 Gbp of data. Genet. Biol. 2010, 11, R62. [Google Scholar]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbob, P.; Nayirc, A.; Bakkaloğlud, A.; Özend, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genet. Res. 2009, 19, 1316–1323. [Google Scholar]
- Mouse Genome Project. Available online: http://www.sanger.ac.uk/resources/mouse/genomes/ (accessed on 28 August 2014).
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar]
- Pipeline Space Home. Available online: https://biowiki.atlassian.net/wiki/display/PS/Pipeline+Space+Home (accessed on 28 August 2014).
- GATK Best Practices. Available online: http://www.broadinstitute.org/gatk/guide/best-practices (accessed on 8 August 2014).
- Lohse, M.; Bolger, A.M.; Nagel, A.; Fernie, A.R.; Lunn, J.E.; Stitt, M.; Usadel, B. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucl. Acids Res. 2012, 40, W622–W627. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows—Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Br. Bioinform. 2010, 11, 473–483. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Skliris, G.P.; Rowan, B.G.; Al-Dhaheri, M.; Williams, C.; Troup, S.; Begic, S.; Parisien, M.; Watson, P.H.; Murphy, L.C. Immunohistochemical validation of multiple phospho-specific epitopes for estrogen receptor alpha (ERalpha) in tissue microarrays of ERalpha positive human breast carcinomas. Breast Cancer Res. Treat 2009, 118, 443–453. [Google Scholar]
- Picard. Available online: http://picard.sourceforge.net (accessed on 28 August 2014).
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. Available online: http://arxiv.org/abs/1207.3907 (accessed on 28 August 2014).
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar]
- Moresco, E.M.; Li, X.; Beutler, B. Going forward with genetics: Recent technological advances and forward genetics in mice. Am. J. Pathol. 2013, 182, 1462–1473. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucl. Acids Res. 2001, 29, 308–311. [Google Scholar]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar]
- Kwiatkowski, D.P. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 2005, 77, 171–192. [Google Scholar]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar]
- Friedman, M.J. Erythrocytic mechanism of sickle cell resistance to malaria. Proc. Natl. Acad. Sci. USA 1978, 75, 1994–1997. [Google Scholar]
- Allen, S.J.; O’Donnell, A.; Alexander, N.D.; Mgone, C.S.; Peto, T.E.; Clegg, J.B.; Alpers, M.P.; Weatherall, D.J. Prevention of cerebral malaria in children in Papua New Guinea by southeast Asian ovalocytosis band 3. Am. J. Trop. Med. Hyg. 1999, 60, 1056–1060. [Google Scholar]
- Foo, L.C.; Rekhraj, V.; Chiang, G.L.; Mak, J.W. Ovalocytosis protects against severe malaria parasitemia in the Malayan aborigines. Am. J. Trop. Med. Hyg. 1992, 47, 271–275. [Google Scholar]
- Genton, B.; al-Yaman, F.; Mgone, C.S.; Alexander, N.; Paniu, M.M.; Alpers, M.P.; Mokela, D. Ovalocytosis and cerebral malaria. Nature 1995, 378, 564–565. [Google Scholar]
- Miller, L.H.; Mason, S.J.; Clyde, D.F.; McGinniss, M.H. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 1976, 295, 302–304. [Google Scholar]
- Ruwende, C.; Khoo, S.C.; Snow, R.W.; Yates, S.N.; Kwiatkowski, D.; Gupta, S.; Warn, P.; Allsopp, C.E.; Gilbert, S.C.; Peschu, N.; et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature 1995, 376, 246–249. [Google Scholar]
- Tishkoff, S.A.; Varkonyi, R.; Cahinhinan, N.; Abbes, S.; Argyropoulos, G.; Destro-Bisol, G.; Drousiotou, A.; Dangerfield, B.; Lefranc, G.; Loiselet, J.; et al. Haplotype diversity and linkage disequilibrium at human G6PD: Recent origin of alleles that confer malarial resistance. Science 2001, 293, 455–462. [Google Scholar]
- Aitman, T.J.; Cooper, L.D.; Norsworthy, P.J.; Wahid, F.N.; Gray, J.K.; Curtis, B.R.; McKeigue, P.M.; Kwiatkowski, D.; Greenwood, B.M.; Snow, R.W.; et al. Malaria susceptibility and CD36 mutation. Nature 2000, 405, 1015–1016. [Google Scholar]
- Omi, K.; Ohashi, J.; Patarapotikul, J.; Hananantachai, H.; Naka, I.; Pottere, S.; Medanad, I.M.; Miua, J.; Ball, H.J. CD36 polymorphism is associated with protection from cerebral malaria. Am. J. Hum. Genet. 2003, 72, 364–374. [Google Scholar]
- Hill, A.V.; Allsopp, C.E.; Kwiatkowski, D.; Anstey, N.M.; Twumasi, P.; Rowe, P.A.; Bennett, S.; Brewster, D.; McMichael, A.J.; Greenwood, B.M. Common west African HLA antigens are associated with protection from severe malaria. Nature 1991, 352, 595–600. [Google Scholar]
- McGuire, W.; Hill, A.V.; Allsopp, C.E.; Greenwood, B.M.; Kwiatkowski, D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 1994, 371, 508–510. [Google Scholar]
- Bongfen, S.E.; Laroque, A.; Berghout, J.; Gros, P. Genetic and genomic analyses of host-pathogen interactions in malaria. Trends Parasitol. 2009, 25, 417–422. [Google Scholar]
- Verra, F.; Mangano, V.D.; Modiano, D. Genetics of susceptibility to Plasmodium falciparum: From classical malaria resistance genes towards genome-wide association studies. Parasit. Immunol. 2009, 31, 234–253. [Google Scholar]
- Fortin, A.; Stevenson, M.M.; Gros, P. Complex genetic control of susceptibility to malaria in mice. Genes Immun. 2002, 3, 177–186. [Google Scholar]
- Hunt, N.H.; Golenser, J.; Chan-Ling, T.; Parekh, S.; Rae, C.; Pottere, S.; Medanad, I.M.; Miua, J.; Ball, H.J. Immunopathogenesis of cerebral malaria. Int. J. Parasitol. 2006, 36, 569–582. [Google Scholar]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar]
- Newton, C.R.; Hien, T.T.; White, N. Cerebral malaria. J. Neurol. Neurosurg. Psychiatry 2000, 69, 433–441. [Google Scholar]
- Tripathi, A.K.; Sha, W.; Shulaev, V.; Stins, M.F.; Sullivan, D.J., Jr. Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood 2009, 114, 4243–4252. [Google Scholar]
- Brown, H.; Hien, T.T.; Day, N.; Mai, N.T.; Chuong, L.V.; Chau, T.T.; Loc, P.P.; Phu, N.H.; Bethell, D.; Farrar, J.; et al. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol. Appl. Neurobiol. 1999, 25, 331–340. [Google Scholar]
- Mishra, S.K.; Newton, C.R. Diagnosis and management of the neurological complications of falciparum malaria. Nat. Rev. Neurol. 2009, 5, 189–198. [Google Scholar]
- Hafalla, J.C.; Claser, C.; Couper, K.N.; Grau, G.E.; Renia, L.; de Souza, J.B.; Riley, E.M. The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology. PLoS Pathog. 2012, 8, e1002504. [Google Scholar]
- Lacerda-Queiroz, N.; Rodrigues, D.H.; Vilela, M.C.; Rachid, M.A.; Soriani, F.M.; Sousa, L.P.; Campos, R.D.; Quesniaux, V.F.; Teixeira, M.M.; Teixeira, A.L. Platelet-activating factor receptor is essential for the development of experimental cerebral malaria. Am. J. Pathol. 2012, 180, 246–255. [Google Scholar]
- Lou, J.; Lucas, R.; Grau, G.E. Pathogenesis of cerebral malaria: Recent experimental data and possible applications for humans. Clin. Microbiol. Rev. 2001, 14, 810–820. [Google Scholar]
- De Souza, J.B.; Riley, E.M. Cerebral malaria: The contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 2002, 4, 291–300. [Google Scholar]
- Senaldi, G.; Vesin, C.; Chang, R.; Grau, G.E.; Piguet, P.F. Role of polymorphonuclear neutrophil leukocytes and their integrin CD11a (LFA-1) in the pathogenesis of severe murine malaria. Infect. Immun. 1994, 62, 1144–1149. [Google Scholar]
- Bagot, S.; Idrissa Boubou, M.; Campinom, S.; Behrschmidt, C.; Gorgette, O.; Guénet, J.L.; Penha-Gonçalves, C.; Mazier, D.; Pied, S.; Cazenave, P.A. Susceptibility to experimental cerebral malaria induced by Plasmodium berghei ANKA in inbred mouse strains recently derived from wild stock. Infect. Immun. 2002, 70, 2049–2056. [Google Scholar]
- Berghout, J.; Min-Oo, G.; Tam, M.; Gauthier, S.; Stevenson, M.M.; Gros, P. Identification of a novel cerebral malaria susceptibility locus (Berr5) on mouse chromosome 19. Genes Immun. 2010, 11, 310–318. [Google Scholar]
- Bopp, S.E.; Rodrigo, E.; Gonzalez-Paez, G.E.; Frazer, M.; Barnes, S.W.; Valim, C.; Watson, J.; Walker, J.R.; Schmedt, C.; Winzeler, E.A. Identification of the Plasmodium berghei resistance locus 9 linked to survival on chromosome 9. Malar. J. 2013, 12, 316. [Google Scholar]
- Campino, S.; Bagot, S.; Bergman, M.L.; Almeida, P.; Sepulveda, N.; Pied, S.; Penha-Gonçalves, C.; Holmberg, D.; Cazenave, P.-A. Genetic control of parasite clearance leads to resistance to Plasmodium berghei ANKA infection and confers immunity. Genes Immun. 2005, 6, 416–421. [Google Scholar]
- Ohno, T.; Nishimura, M. Detection of a new cerebral malaria susceptibility locus, using CBA mice. Immunogenetics 2004, 56, 675–678. [Google Scholar]
- Bopp, S.E.; Ramachandran, V.; Henson, K.; Luzader, A.; Lindstrom, M.; Spooner, M.; Steffy, B.M.; Suzuki, O.; Janse, C.; Waters, A.P.; et al. Genome wide analysis of inbred mouse lines identifies a locus containing Ppar-gamma as contributing to enhanced malaria survival. PLoS ONE 2010, 5, e10903. [Google Scholar]
- Bongfen, S.E.; Rodrigue-Gervais, I.G.; Berghout, J.; Torre, S.; Cingolani, P.; Wiltshire, S.A.; Leiva-Torres, G.A.; Letourneau, L.; Sladek, R.; Blanchette, M.; et al. An N-ethyl-N-nitrosourea (ENU)-induced dominant negative mutation in the JAK3 kinase protects against cerebral malaria. PLoS ONE 2012, 7, e31012. [Google Scholar]
- Nosaka, T.; van Deursen, J.M.; Tripp, R.A.; Thierfelder, W.E.; Witthuhn, B.A.; McMickle, A.P.; Doherty, P.C.; Grosveld, G.C.; Ihle, J.N. Defective lymphoid development in mice lacking Jak3. Science 1995, 270, 800–802. [Google Scholar]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. The genetic association of variants in CD6, TNFRSF1A and IRF8 to multiple sclerosis: A multicenter case-control study. PLoS ONE 2011, 6, e18813. [Google Scholar]
- Jakkula, E.; Leppa, V.; Sulonen, A.M.; Varilo, T.; Kallio, S.; Kemppinen, A.; Purcell, S.; Koivisto, K.; Tienari, P.; Sumelahti, M.; et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 2010, 86, 285–291. [Google Scholar]
- Torre, S.; van Bruggen, R.; Kennedy, J.M.; Berghout, J.; Bongfen, S.E.; Langat, P.; Lathrop, M.; Vidal, S.M.; Gros, P. Susceptibility to lethal cerebral malaria is regulated by epistatic interaction between chromosome 4 (Berr6) and chromosome 1 (Berr7) loci in mice. Genes Immun. 2013, 14, 470. [Google Scholar]
- Torre, S.; Gros, P.; Department of Human Genetics, Department of Biochemistry, and Complex Traits Group, McGill University, Montréal, QC, Canada. Unpublished data. 2014.
- Pignata, C.; Fusco, A.; Amorosi, S. Human clinical phenotype associated with FOXN1 mutations. Adv. Exp. Med. Biol. 2009, 665, 195–206. [Google Scholar]
- Rank, G.; Sutton, R.; Marshall, V.; Lundie, R.J.; Caddy, J.; Romeo, T.; Fernandez, K.; Mccormack, M.P.; Cooke, B.M.; Foote, S.J.; et al. Novel roles for erythroid Ankyrin-1 revealed through an ENU-induced null mouse mutant. Blood 2009, 113, 3352–3362. [Google Scholar]
- Greth, A.; Lampkin, S.; Mayura-Guru, P.; Rodda, F.; Drysdale, K.; Roberts-Thomson, M.; McMorran, B.J.; Foote, S.J.; Burgio, G. A novel ENU-mutation in ankyrin-1 disrupts malaria parasite maturation in red blood cells of mice. PLoS One 2012, 7, e38999. [Google Scholar]
- Belnoue, E.; Kayibanda, M.; Vigario, A.M.; Deschemin, J.C.; van Rooijen, N.; Viguier, M.; Snounou, G.; Rénia, L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 2002, 169, 6369–6375. [Google Scholar]
- Finley, R.W.; Mackey, L.J.; Lambert, P.H.; Virulent, P. berghei malaria: Prolonged survival and decreased cerebral pathology in cell-dependent nude mice. J. Immunol. 1982, 129, 2213–2218. [Google Scholar]
- Renia, L.; Potter, S.M.; Mauduit, M.; Rosa, D.S.; Kayibanda, M.; Deschemina, J.; Snounoub, G.; Grüner, A.C. Pathogenic T cells in cerebral malaria. Int. J. Parasitol. 2006, 36, 547–554. [Google Scholar]
- Grau, G.E.; Piguet, P.F.; Engers, H.D.; Louis, J.A.; Vassalli, P.; Lambert, P.H. L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J. Immunol. 1986, 137, 2348–2354. [Google Scholar]
- Crump, J.A.; Luby, S.P.; Mintz, E.D. The global burden of typhoid fever. Bull. World Health Organ. 2004, 82, 346–353. [Google Scholar]
- Mastroeni, P.; Maskell, D. Salmonella Infections: Clinical, Immunological, and Molecular Aspects; Cambridge University Press: Cambridge, UK, 2006; Volume 13, p. 381. [Google Scholar]
- Gordon, M.A.; Banda, H.T.; Gondwe, M.; Gordon, S.B.; Boeree, M.J.; Walsh, A.L.; Corkill, J.E.; Hart, C.A.; Gilks, C.F.; Molyneux, M.E. Non-typhoidal Salmonella bacteraemia among HIV-infected Malawian adults: High mortality and frequent recrudescence. AIDS 2002, 16, 1633–1641. [Google Scholar]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M. The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar]
- Mittrucker, H.W.; Kaufmann, S.H. Immune response to infection with Salmonella typhimurium in mice. J. Leukoc. Biol. 2000, 67, 457–463. [Google Scholar]
- Dougan, G.; John, V.; Palmer, S.; Mastroeni, P. Immunity to salmonellosis. Immunol. Rev. 2011, 240, 196–210. [Google Scholar]
- Alcais, A.; Abel, L.; Casanova, J.L. Human genetics of infectious diseases: Between proof of principle and paradigm. J. Clin. Invest. 2009, 119, 2506–2514. [Google Scholar]
- Bustamante, J.; Zhang, S.Y.; von Bernuth, H.; Abel, L.; Casanova, J.L. From infectious diseases to primary immunodeficiencies. Immunol. Allergy Clin. North Am. 2008, 28, 235–258. [Google Scholar]
- Casanova, J.L.; Fieschi, C.; Zhang, S.Y.; Abel, L. Revisiting human primary immunodeficiencies. J. Intern. Med. 2008, 264, 115–127. [Google Scholar]
- De Beaucoudrey, L.; Samarina, A.; Bustamante, J.; Cobat, A.; Boisson-Dupuis, S.; Feinberg, J.; Al-Muhsen, S.; Jannière, L.; Rose, Y.; Desurenaim, M.; et al. Revisiting human IL-12Rbeta1 deficiency: A survey of 141 patients from 30 countries. Med. (Baltim.) 2010, 89, 381–402. [Google Scholar]
- Gordon, M. Salmonella infections in immunocompromised adults. J. Infect. 2008, 56, 413–422. [Google Scholar]
- Lammas, D.A.; Casanova, J.L.; Kumararatne, D.S. Clinical consequences of defects in the IL-12-dependent interferon-gamma (IFN-gamma) pathway. Clin. Exp. Immunol. 2000, 121, 417–425. [Google Scholar]
- Dunstan, S.J.; Stephens, H.A.; Blackwell, J.M.; Duc, C.M.; Lanh, M.N.; Dudbridge, F.; Phuong, C.X.; Luxemburger, C.; Wain, J.; Ho, V.A.; et al. Genes of the class II and class III major histocompatibility complex are associated with typhoid fever in Vietnam. J. Infect. Dis. 2001, 183, 261–268. [Google Scholar]
- House, D.; Bishop, A.; Parry, C.; Dougan, G.; Wain, J. Typhoid fever: Pathogenesis and disease. Curr. Opin. Infect. Dis. 2001, 14, 573–578. [Google Scholar]
- Santos, R.L.; Zhang, S.; Tsolis, R.M.; Kingsley, R.A.; Adams, L.G.; Bäumler, A.J. Animal models of Salmonella infections: Enteritis versus typhoid fever. Microbes Infect. 2001, 3, 1335–1344. [Google Scholar]
- Roy, M.F.; Malo, D. Genetic regulation of host responses to Salmonella infection in mice. Genes Immun. 2002, 3, 381–393. [Google Scholar]
- Malo, D.; Vogan, K.; Vidal, S.; Hu, J.; Cellier, M.; Schurr, E.; Fuks, A.; Bumstead, N.; Morgan, K.; Gros, P. Haplotype mapping and sequence analysis of the mouse Nramp gene predict susceptibility to infection with intracellular parasites. Genomics 1994, 23, 51–61. [Google Scholar]
- Qureshi, S.T.; Lariviere, L.; Leveque, G.; Clermont, S.; Moore, K.J.; Gros, P.; Malo, D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 1999, 189, 615–625. [Google Scholar]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Eefective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar]
- Vidal, S.M.; Malo, D.; Vogan, K.; Skamene, E.; Gros, P. Natural resistance to infection with intracellular parasites: Isolation of a candidate for Bcg. Cell 1993, 73, 469–485. [Google Scholar]
- Roy, M.F.; Riendeau, N.; Bedard, C.; Helie, P.; Min-Oo, G.; Turcotte, K.; Gros, P.; Canonne-Hergaux, F.; Malo, D. Pyruvate kinase deficiency confers susceptibility to Salmonella typhimurium infection in mice. J. Exp. Med. 2007, 204, 2949–2961. [Google Scholar]
- Yuki, K.E.; Eva, M.M.; Richer, E.; Chung, D.; Paquet, M.; Cellier, M.; Canonne-Hergaux, F.; Vaulont, S.; Vidal, S.M.; Malo, D.; et al. Suppression of hepcidin expression and iron overload mediate Salmonella susceptibility in ankyrin 1 ENU-induced mutant. PLoS One 2013, 8, e55331. [Google Scholar]
- Eva, M.M.; Yuki, K.E.; Dauphinee, S.M.; Schwartzentruber, J.A.; Pyzik, M.; Paquet, M.; Lathrop, M.; Majewski, J.; Vidal, S.M.; Malo, D.; et al. Altered IFN-gamma-mediated immunity and transcriptional expression patterns in N-Ethyl-N-nitrosourea-induced STAT4 mutants confer susceptibility to acute typhoid-like disease. J. Immunol. 2014, 192, 259–270. [Google Scholar]
- Richer, E.; Prendergast, C.; Zhang, D.E.; Qureshi, S.T.; Vidal, S.M.; Malo, D. N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-alphass signaling and suppresses STAT4-induced IFN-gamma production, resulting in increased susceptibility to Salmonella typhimuriu. J. Immunol. 2010, 185, 3593–3601. [Google Scholar]
- Malakhova, O.; Malakhov, M.; Hetherington, C.; Zhang, D.E. Lipopolysaccharide activates the expression of ISG15-specific protease UBP43 via interferon regulatory factor 3. J. Biol. Chem. 2002, 277, 14703–14711. [Google Scholar]
- Kim, K.I.; Yan, M.; Malakhova, O.; Luo, J.K.; Shen, M.F.; Zou, W.; de la Torre, J.C.; Zhang, D. Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling. Mol. Cell. Biol. 2006, 26, 472–479. [Google Scholar]
- Richer, E.; Yuki, K.E.; Dauphinee, S.M.; Lariviere, L.; Paquet, M.; Malo, D. Impact of Usp18 and IFN signaling in Salmonella-induced typhlitis. Genes Immun. 2011, 12, 531–543. [Google Scholar]
- Dauphinee, S.M.; Richer, E.; Eva, M.M.; McIntosh, F.; Paquet, M.; Dangoor, D.; Burkart, C.; Zhang, D.E.; Gruenheid, S.; Gros, P.; et al. Contribution of increased ISG15, ISGylation and deregulated type I IFN signaling in Usp18 mutant mice during the course of bacterial infections. Genes Immun. 2014, 15, 282–292. [Google Scholar]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar]
- Eber, S.W.; Gonzalez, J.M.; Lux, M.L.; Scarpa, A.L.; Tse, W.T.; Dornwell, M.; Herbers, J.; Kugler, W.; Ozcan, R.; Pekrun, A.; et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat. Genet. 1996, 13, 214–218. [Google Scholar]
- Min-Oo, G.; Fortin, A.; Tam, M.F.; Nantel, A.; Stevenson, M.M.; Gros, P. Pyruvate kinase deficiency in mice protects against malaria. Nat. Genet. 2003, 35, 357–362. [Google Scholar]
- Cunnington, A.J.; de Souza, J.B.; Walther, M.; Riley, E.M. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 2012, 18, 120–127. [Google Scholar]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shame, L.; Hartung, T.; Zähringer, U.; et al. CD36 is a sensor of diacylglycerides. Nature 2005, 433, 523–527. [Google Scholar]
- Xiao, N.; Eidenschenk, C.; Krebs, P.; Brandl, K.; Blasius, A.L.; Xia, Y.; Khovananth, K.; Smart, N.G.; Beutler, B. The Tpl2 mutation Sluggish impairs type I IFN production and increases susceptibility to group B streptococcal disease. J. Immunol. 2009, 183, 7975–7983. [Google Scholar]
- Rutschmann, S.; Hoebe, K.; Zalevsky, J.; Du, X.; Mann, N.; Dahiyat, B.I.; Steed, P.; Beutler, B. PanR1, a dominant negative missense allele of the gene encoding TNF-alpha (Tnf), does not impair lymphoid development. J. Immunol. 2006, 176, 7525–7532. [Google Scholar]
- Sauer, J.D.; Sotelo-Troha, K.; von Moltke, J.; Monroe, K.M.; Rae, C.S.; Brubaker, S.W.; Hyodo, M.; Hayakawa, Y.; Woodward, J.J.; Portnoy, D.A.; et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 2011, 79, 688–694. [Google Scholar]
- Berger, M.; Krebs, P.; Crozat, K.; Li, X.; Croker, B.A.; Siggs, O.M.; Popkin, D.; Du, X.; Lawson, B.R.; Theofilopoulos, A.N.; et al. An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nat. Immunol. 2010, 11, 335–343. [Google Scholar]
- Ordonez-Rueda, D.; Jonsson, F.; Mancardi, D.A.; Zhao, W.; Malzac, A.; Liang, Y.; Bertosio, E.; Grenot, P.; Blanquet, V.; Sabrautzki, S.; et al. A hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia. Eur. J. Immunol. 2012, 42, 2395–2408. [Google Scholar]
- Georgel, P.; Crozat, K.; Lauth, X.; Makrantonaki, E.; Seltmann, H.; Sovath, S.; Hoebe, K.; Du, X.; Rutschmann, S.; Jiang, Z.; et al. A toll-like receptor 2-responsive lipid effector pathway protects mammals against skin infections with gram-positive bacteria. Infect. Immun. 2005, 73, 4512–4521. [Google Scholar]
- Croker, B.A.; Lawson, B.R.; Rutschmann, S.; Berger, M.; Eidenschenk, C.; Blasius, A.L.; Moresco, E.M.Y.; Sovath, S.; Cengia, L.; Shultz, L.D.; et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc. Natl. Acad. Sci. USA 2008, 105, 15028–15033. [Google Scholar]
- Jaeger, B.N.; Donadieu, J.; Cognet, C.; Bernat, C.; Ordonez-Rueda, D.; Barlogis, V.; Mahlaoui, N.; Fenis, A.; Narni-Mancinelli, E.; Beaupain, B.; et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J. Exp. Med. 2012, 209, 565–580. [Google Scholar]
- Barquero-Calvo, E.; Martirosyan, A.; Ordonez-Rueda, D.; Arce-Gorvel, V.; Alfaro-Alarcon, A.; Lepidi, H.; Malissen, B.; Malissen, M.; Gorvel, J.; Moreno, E. Neutrophils exert a suppressive effect on Th1 responses to intracellular pathogen Brucella abortus. PLoS Pathog. 2013, 9, e1003167. [Google Scholar]
- Krmpotic, A.; Bubic, I.; Polic, B.; Lucin, P.; Jonjic, S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003, 5, 1263–1277. [Google Scholar]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar]
- Pyzik, M.; Gendron-Pontbriand, E.M.; Vidal, S.M. The impact of Ly49-NK cell-dependent recognition of MCMV infection on innate and adaptive immune responses. J. Biomed. Biotechnol. 2011, 2011, 641702. [Google Scholar]
- Moresco, E.M.; Beutler, B. Resisting viral infection: The gene by gene approach. Curr. Opin. Virol. 2011, 1, 513–518. [Google Scholar]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3516–3521. [Google Scholar]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; George, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar]
- Crozat, K.; Georgel, P.; Rutschmann, S.; Mann, N.; Du, X.; Hoebe, K.; Beutlerm, B. Analysis of the MCMV resistome by ENU mutagenesis. Mamm. Genome 2006, 17, 398–406. [Google Scholar]
- Siggs, O.M.; Berger, M.; Krebs, P.; Arnold, C.N.; Eidenschenk, C.; Huberb, C.; Piriea, E.; Smarta, N.G.; Khovanantha, K.; Xia, Y.; et al. A mutation of Ikbkg causes immune deficiency without impairing degradation of IkappaB alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 3046–3051. [Google Scholar]
- Won, S.; Eidenschenk, C.; Arnold, C.N.; Siggs, O.M.; Sun, L.; Brandla, K.; Mullenb, T.; Nemerowb, G.R.; Morescoa, E.M.Y.; Beutler, B. Increased susceptibility to DNA virus infection in mice with a GCN2 mutation. J. Virol. 2012, 86, 1802–1808. [Google Scholar]
- Biron, C.A. Initial and innate responses to viral infections—Pattern setting in immunity or disease. Curr. Opin. Microbiol. 1999, 2, 374–381. [Google Scholar]
- Bukowski, J.F.; Woda, B.A.; Habu, S.; Okumura, K.; Welsh, R.M. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immunol. 1983, 131, 1531–1538. [Google Scholar]
- Bukowski, J.F.; Woda, B.A.; Welsh, R.M. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J. Virol. 1984, 52, 119–128. [Google Scholar]
- Welsh, R.M.; Dundon, P.L.; Eynon, E.E.; Brubaker, J.O.; Koo, G.C.; O’Donnell, C.L. Demonstration of the antiviral role of natural killer cells in vivo with a natural killer cell-specific monoclonal antibody (NK 1.1). Nat. Immun. Cell Growth Regul. 1990, 9, 112–120. [Google Scholar]
- Brown, M.G.; Dokun, A.O.; Heusel, J.W.; Smith, H.R.; Beckman, D.L.; Blattenberger, E.A.; Dubbelde, C.E.; Stone, L.R.; Scalzo, A.A.; Yokoyama, W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001, 292, 934–937. [Google Scholar]
- Lee, S.H.; Girard, S.; Macina, D.; Busa, M.; Zafer, A.; Belouchi, A.; Gros, P.; Vidal, S.M. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 2001, 28, 42–45. [Google Scholar]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar]
- Smith, H.R.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; et al. Recognition of a virus-encodedligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 8826–8831. [Google Scholar]
- Dokun, A.O.; Kim, S.; Smith, H.R.; Kang, H.S.; Chu, D.T.; Yokoyama, W.M. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2001, 2, 951–956. [Google Scholar]
- Barnes, M.J.; Aksoylar, H.; Krebs, P.; Bourdeau, T.; Arnold, C.N.; Xia, Y.; Khovananth, K.; Engel, I.; Sovath, S.; Lampe, K.; et al. Loss of T cell and B cell quiescence precedes the onset of microbial flora-dependent wasting disease and intestinal inflammation in Gimap5-deficient mice. J. Immunol. 2010, 184, 3743–3754. [Google Scholar]
- Crozat, K.; Hoebe, K.; Ugolini, S.; Hong, N.A.; Janssen, E.; Rutschmann, S.; Mudd, S.; Sovath, S.; Vivier, E.; Beutler, B. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: A mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J. Exp. Med. 2007, 204, 853–863. [Google Scholar]
- Crozat, K.; Eidenschenk, C.; Jaeger, B.N.; Krebs, P.; Guia, S.; Beutler, B.; Vivier, E.; Ugolini, S. Impact of beta2 integrin deficiency on mouse natural killer cell development and function. Blood 2011, 117, 2874–2882. [Google Scholar]
- Eidenschenk, C.; Crozat, K.; Krebs, P.; Arens, R.; Popkin, D.; Arnolda, C.N.; Blasiusa, A.L.; Benedictb, C.A.; Morescoa, E.M.Y.; Xiaa, Y.; et al. Flt3 permits survival during infection by rendering dendritic cells competent to activate NK cells. Proc. Natl. Acad. Sci. USA 2010, 107, 9759–9764. [Google Scholar]
- Croker, B.; Crozat, K.; Berger, M.; Xia, Y.; Sovath, S.; Schaffer, L.; Eleftherianos, I.; Imler, J.; Beutler, B. ATP-sensitive potassium channels mediate survival during infection in mammals and insects. Nat. Genet. 2007, 39, 1453–1460. [Google Scholar]
- Abel, L.; Plancoulaine, S.; Jouanguy, E.; Zhang, S.Y.; Mahfoufi, N.; Nicolas, N.; Sancho-Shimizu, V.; Alcaïs, A.; Guo, Y.; Cardon, A.; et al. Age-dependent Mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J. Pediatr. 2010, 157, 623–629. [Google Scholar]
- Yao, H.W.; Ling, P.; Chen, S.H.; Tung, Y.Y.; Chen, S.H. Factors affecting herpes simplex virus reactivation from the explanted mouse brain. Virology 2012, 433, 116–123. [Google Scholar]
- Kennedy, P.G.; Chaudhuri, A. Herpes simplex encephalitis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 237–238. [Google Scholar]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar]
- Sancho-Shimizu, V.; Perez de Diego, R.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Invest. 2011, 121, 4889–4902. [Google Scholar]
- Perez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar]
- Herman, M.; Ciancanelli, M.; Ou, Y.H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; de Diego, R.P.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar]
- Xia, Y.; Won, S.; Du, X.; Lin, P.; Ross, C.; la Vine, D.; Wiltshire, S.; Leiva, G.; Vidal, S.M.; Whittle, B.; et al. Bulk segregation mapping of mutations in closely related strains of mice. Genetics 2010, 186, 1139–1146. [Google Scholar]
- Caignard, G.; Leiva-Torres, G.A.; Leney-Greene, M.; Charbonneau, B.; Dumaine, A.; Fodil-Cornu, N.; Pyzik, M.; Cingolani, P.; Schwartzentruber, J.; Dupaul-Chicoine, J.; et al. Genome-wide mouse mutagenesis reveals CD45-mediated T cell function as critical in protective immunity to HSV-1. PLoS Pathog. 2013, 9, e1003637. [Google Scholar]
- Caignard, G.; Vidal, S.M.; Department of Human Genetics and Complex Traits Group, McGill University, Montréal, QC, Canada. Unpublished data. 2014.
- Caignard, G.; Gros, P.; Vidal, S.M.; Department of Human Genetics, Department of Biochemistry, and Complex Traits Group, McGill University, Montréal, QC, Canada. Unpublished data. 2014.
- Tchilian, E.Z.; Beverley, P.C. Altered CD45 expression and disease. Trends Immunol. 2006, 27, 146–153. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caignard, G.; Eva, M.M.; Van Bruggen, R.; Eveleigh, R.; Bourque, G.; Malo, D.; Gros, P.; Vidal, S.M. Mouse ENU Mutagenesis to Understand Immunity to Infection: Methods, Selected Examples, and Perspectives. Genes 2014, 5, 887-925. https://doi.org/10.3390/genes5040887
Caignard G, Eva MM, Van Bruggen R, Eveleigh R, Bourque G, Malo D, Gros P, Vidal SM. Mouse ENU Mutagenesis to Understand Immunity to Infection: Methods, Selected Examples, and Perspectives. Genes. 2014; 5(4):887-925. https://doi.org/10.3390/genes5040887
Chicago/Turabian StyleCaignard, Grégory, Megan M. Eva, Rebekah Van Bruggen, Robert Eveleigh, Guillaume Bourque, Danielle Malo, Philippe Gros, and Silvia M. Vidal. 2014. "Mouse ENU Mutagenesis to Understand Immunity to Infection: Methods, Selected Examples, and Perspectives" Genes 5, no. 4: 887-925. https://doi.org/10.3390/genes5040887
APA StyleCaignard, G., Eva, M. M., Van Bruggen, R., Eveleigh, R., Bourque, G., Malo, D., Gros, P., & Vidal, S. M. (2014). Mouse ENU Mutagenesis to Understand Immunity to Infection: Methods, Selected Examples, and Perspectives. Genes, 5(4), 887-925. https://doi.org/10.3390/genes5040887