
Replication-Associated Recombinational Repair: Lessons from Budding Yeast

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
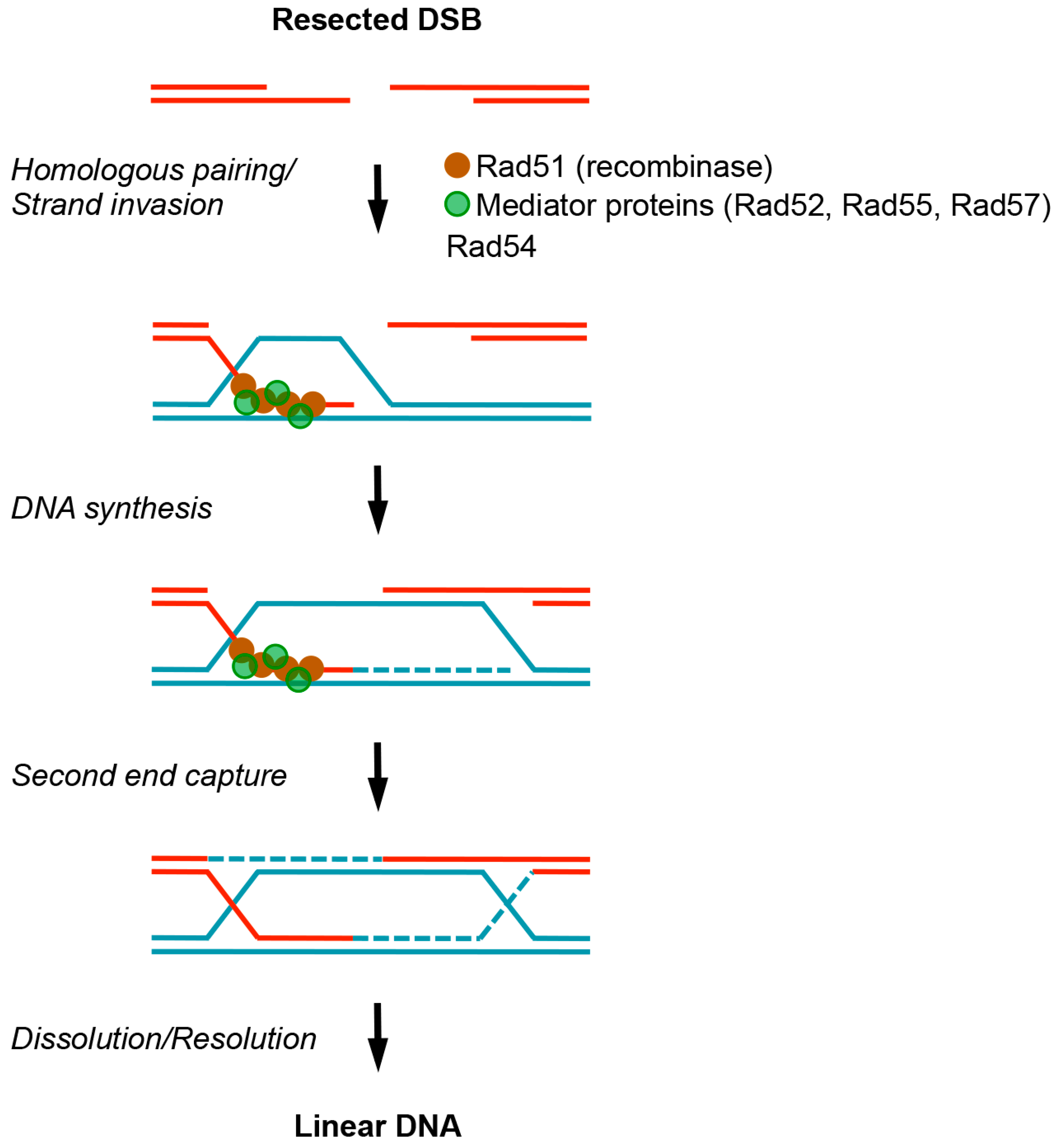
:1. Introduction
2. PCNA Modifications and Their Link to Recombinational Repair
2.1. Readers of PCNA Modifications
2.2. Control of PCNA and Srs2 Levels at Stalled Forks
3. The Shu Complex Promotes Rad51 Function in Recombinational Repair
3.1. Genetic Studies of the Shu Complex
3.2. Mechanisms of Shu Complex Functions
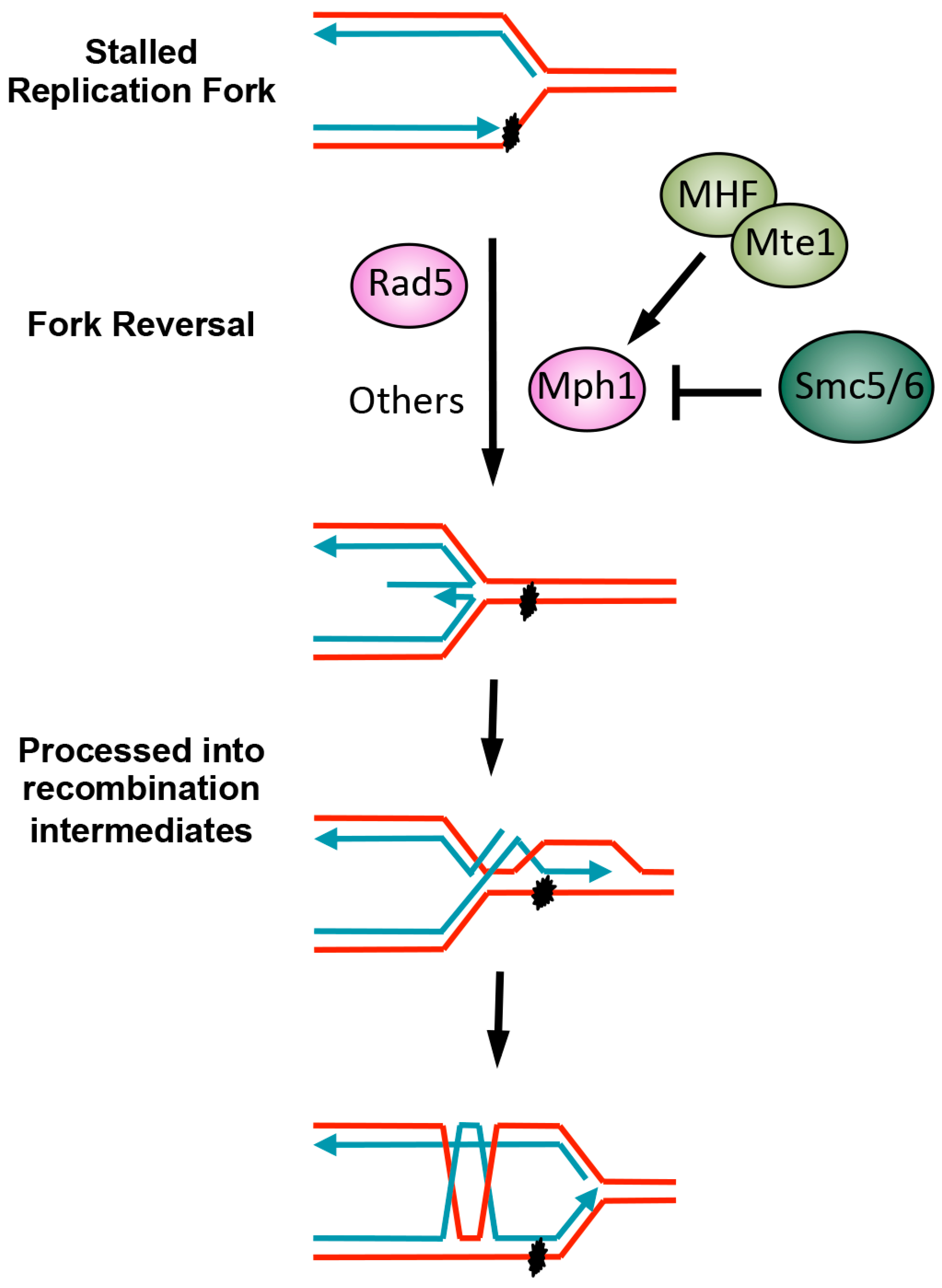
4. Replication Fork Regression by DNA Helicases and Their Regulation
4.1. The Multiple Rad5 Activities
4.2. Mph1 and Its Regulation
4.3. Other DNA Helicases Involved in Fork Regression and Additional Regulators
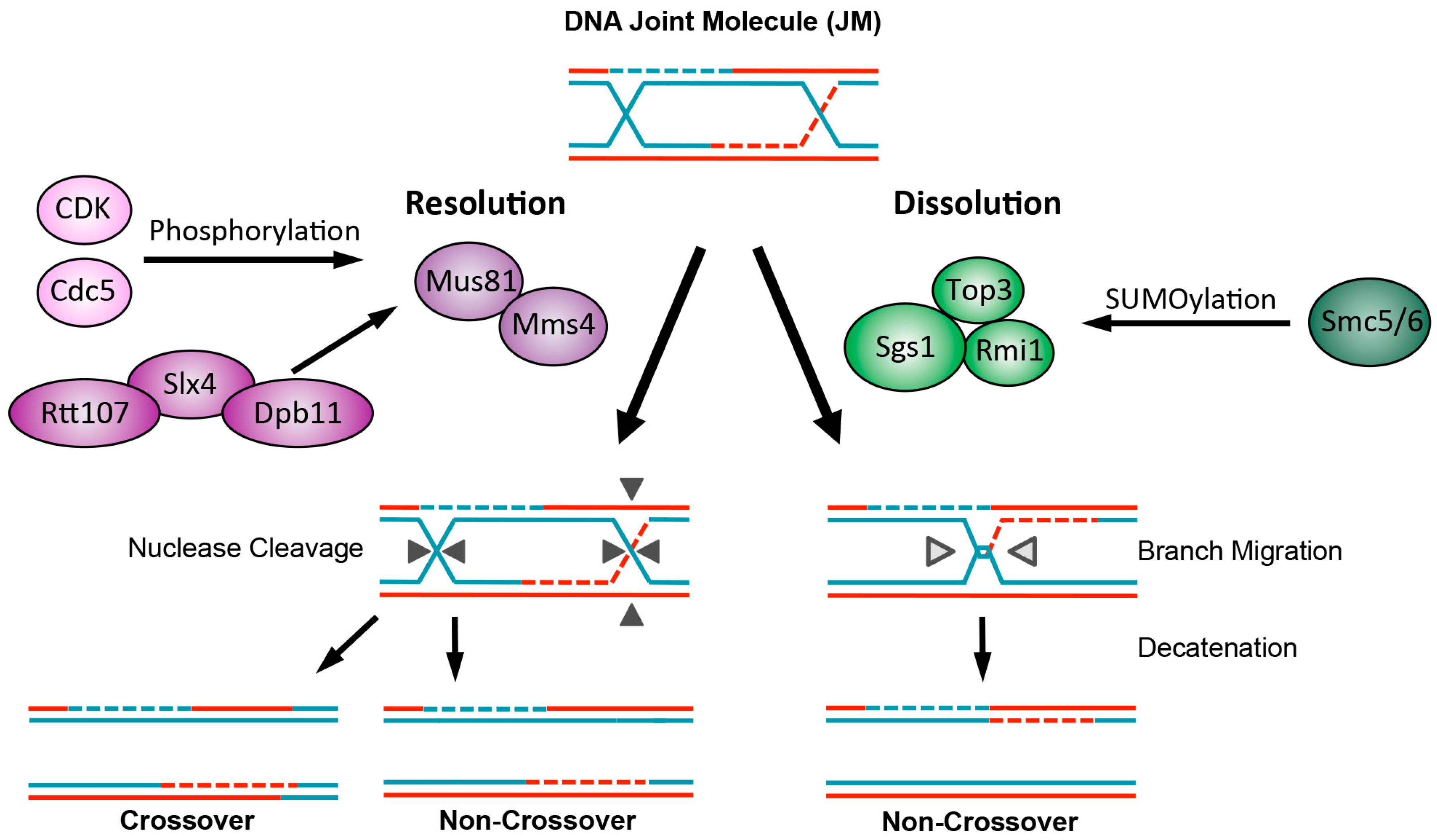
5. Recombination Intermediate Processing Is Controlled by Protein Modifications
5.1. Dissolution by STR Is Regulated by SUMOylation
5.2. Resolution Nucleases Are Regulated by Phosphorylation
6. Roles of Chromatin in Recombinational Repair
6.1. DNA Bending by Hmo1 Promotes Template Switching
6.2. Cohesion Contributes to Efficient Recombinational Repair during Replication
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Replication stress and genome rearrangements: Lessons from yeast models. Curr. Opin. Genet. Dev. 2013, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [PubMed]
- Kowalczykowski, S.C. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Rothstein, R.; Lisby, M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 2014, 198, 795–835. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.D.; Kumar, R.; Srivastava, S.; Ghosh, S.K. Cohesin: Functions beyond sister chromatid cohesion. FEBS Lett. 2013, 587, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D.M. Resolution of recombination intermediates: Mechanisms and regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 027649. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Blackwell, S.; Lin, A.; Li, F.; Qin, Z.; Xiao, W. Error-free DNA-damage tolerance in Saccharomyces cerevisiae. Mutat. Res./Rev. Mutat. Res. 2015, 764, 43–50. [Google Scholar]
- McIntyre, J.; Woodgate, R. Regulation of translesion DNA synthesis: Posttranslational modification of lysine residues in key proteins. DNA Repair 2015, 29, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D. Two-way communications between ubiquitin-like modifiers and DNA. Nat. Struct. Mol. Biol. 2014, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Saugar, I.; Parker, J.L.; Zhao, S.; Ulrich, H.D. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012, 40, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Leuzzi, G.; Marabitti, V.; Pichierri, P.; Franchitto, A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016, 35, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Iwasaki, H.; Ohno, T.; Morishita, T.; Shinagawa, H. A yeast gene, MGS1, encoding a DNA-dependent AAA(+) ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. USA 2001, 98, 8283–8289. [Google Scholar] [CrossRef] [PubMed]
- Vijeh Motlagh, N.D.; Seki, M.; Branzei, D.; Enomoto, T. Mgs1 and Rad18/Rad5/Mms2 are required for survival of Saccharomyces cerevisiae mutants with novel temperature/cold sensitive alleles of the DNA polymerase delta subunit, Pol31. DNA Repair 2006, 5, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Seki, M.; Onoda, F.; Enomoto, T. The product of Saccharomyces cerevisiae WHIP/MGS1, a gene related to replication factor C genes, interacts functionally with DNA polymerase delta. Mol. Genet. Genom. 2002, 268, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Seki, M.; Onoda, F.; Yagi, H.; Kawabe, Y.-I.; Enomoto, T. Characterization of the slow-growth phenotype of S. cerevisiae Whip/Mgs1 Sgs1 double deletion mutants. DNA Repair 2002, 1, 671–682. [Google Scholar] [CrossRef]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [PubMed]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.A.; Mohideen, F.; Lima, C.D. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 2012, 483, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Kolesar, P.; Sarangi, P.; Altmannova, V.; Zhao, X.; Krejci, L. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012, 40, 7831–7843. [Google Scholar] [CrossRef] [PubMed]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef]
- Krejci, L.; van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Sebesta, M.; Sisakova, A.; Plault, N.; Szukacsov, V.; Robert, T.; Pinter, L.; Marini, V.; Kolesar, P.; Haracska, L.; et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013, 32, 742–755. [Google Scholar] [PubMed]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [PubMed]
- Parnas, O.; Zipin-Roitman, A.; Pfander, B.; Liefshitz, B.; Mazor, Y.; Ben-Aroya, S.; Jentsch, S.; Kupiec, M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010, 29, 2611–2622. [Google Scholar] [PubMed]
- Johnson, C.; Gali, V.K.; Takahashi, T.S.; Kubota, T. PCNA retention on DNA into G2/M phase causes genome instability in cells lacking Elg1. Cell Rep. 2016, 16, 684–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urulangodi, M.; Sebesta, M.; Menolfi, D.; Szakal, B.; Sollier, J.; Sisakova, A.; Krejci, L.; Branzei, D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev. 2015, 29, 2067–2080. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Szakal, B.; Chen, Y.-H.; Branzei, D.; Zhao, X. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol. Biol. Cell 2010, 21, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Driscoll, R.; Castellucci, F.; Foiani, M.; Jackson, S.P.; Branzei, D. The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol. Biol. Cell 2009, 20, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Westmoreland, J.W.; Resnick, M.A. Homologous recombination rescues ssDNA gaps generated by nucleotide excision repair and reduced translesion DNA synthesis in yeast G2 cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2895–E2904. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-E.; Rio, A.-G.; Nicolas, A.; Kolodner, R.D. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 11529–11534. [Google Scholar] [CrossRef] [PubMed]
- Shor, E.; Weinstein, J.; Rothstein, R. A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: Four genes involved in error-free DNA repair. Genetics 2005, 169, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Hwang, J.-Y.; Banerjee, S.; Majeed, A.; Gupta, A.; Myung, K. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2004, 101, 9039–9044. [Google Scholar] [CrossRef] [PubMed]
- St. Onge, R.P.; Mani, R.; Oh, J.; Proctor, M.; Fung, E.; Davis, R.W.; Nislow, C.; Roth, F.P.; Giaever, G. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 2007, 39, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Ngo, H.-P.; Hickson, I.D. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol. Biol. Cell 2007, 18, 4062–4073. [Google Scholar] [PubMed]
- Sasanuma, H.; Tawaramoto, M.S.; Lao, J.P.; Hosaka, H.; Sanda, E.; Suzuki, M.; Yamashita, E.; Hunter, N.; Shinohara, M.; Nakagawa, A.; et al. A new protein complex promoting the assembly of Rad51 filaments. Nat. Commun. 2013, 4, 1676. [Google Scholar] [CrossRef] [PubMed]
- She, Z.; Gao, Z.-Q.; Liu, Y.; Wang, W.-J.; Liu, G.-F.; Shtykova, E.V.; Xu, J.-H.; Dong, Y.-H. Structural and SAXS analysis of the budding yeast SHU-complex proteins. FEBS Lett. 2012, 586, 2306–2312. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, X.; Liu, Y.; Ruan, J.; Qi, S.; Niu, L.; Teng, M. Structural analysis of Shu proteins reveals a DNA binding role essential for resisting damage. J. Biol. Chem. 2012, 287, 20231–20239. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.; Wier, A.; Kabbinavar, F.; Bratton-Palmer, D.S.; Ghodke, H.; van Houten, B.; vanDemark, A.P.; Bernstein, K.A. The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic Acids Res. 2013, 41, 4525–4534. [Google Scholar] [CrossRef] [PubMed]
- Gaines, W.A.; Godin, S.K.; Kabbinavar, F.F.; Rao, T.; VanDemark, A.P.; Sung, P.; Bernstein, K.A. Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat. Commun. 2015, 6, 7834. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ball, L.; Chen, W.; Tian, X.; Lambrecht, A.; Hanna, M.; Xiao, W. The yeast Shu complex utilizes homologous recombination machinery for error-free lesion bypass via physical interaction with a Rad51 paralogue. PLoS ONE 2013, 8, e81371. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Zhang, Z.; Herken, B.W.; Westmoreland, J.W.; Lee, A.G.; Mihalevic, M.J.; Yu, Z.; Sobol, R.W.; Resnick, M.A.; Bernstein, K.A. The Shu complex promotes error-free tolerance of alkylation-induced base excision repair products. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Chiba, T.; Ozawa, R.; Yoshida, M.; Hattori, M.; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Chahwan, C.; Gao, H.; Blais, V.; Wohlschlegel, J.; Yates, J.R.; Mcgowan, C.H.; Russell, P. Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J. 2006, 25, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.A.; Reid, R.J.D.; Sunjevaric, I.; Demuth, K.; Burgess, R.C.; Rothstein, R. The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase. Mol. Biol. Cell 2011, 22, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- McClendon, T.B.; Sullivan, M.R.; Bernstein, K.A.; Yanowitz, J.L. Promotion of Homologous Recombination by SWS-1 in Complex with RAD-51 Paralogs in Caenorhabditis elegans. Genetics 2016, 203, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Špírek, M.; Chaurasiya, K.R.; Ward, J.D.; Carzaniga, R.; Yu, X.; Egelman, E.H.; Collinson, L.M.; Rueda, D.; Krejci, L.; et al. Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell 2015, 162, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.G.; Zhang, K.; Cobb, J.A.; Boone, C.; Xiao, W. The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol. Microbiol. 2009, 73, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.G.; Xu, X.; Blackwell, S.; Hanna, M.D.; Lambrecht, A.D.; Xiao, W. The Rad5 helicase activity is dispensable for error-free DNA post-replication repair. DNA Repair 2014, 16, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Batke, S.; Szakal, B.; Lowther, J.; Hao, F.; Sarangi, P.; Branzei, D.; Ulrich, H.D.; Zhao, X. Concerted and differential actions of two enzymatic domains underlie Rad5 contributions to DNA damage tolerance. Nucleic Acids Res. 2015, 43, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Minca, E.C.; Kowalski, D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol. Cell 2010, 38, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Kou, H.; Xie, Z.; Zhou, Y.; Feng, X.; Wang, L.; Wang, Z. A non-catalytic function of Rev1 in translesion DNA synthesis and mutagenesis is mediated by its stable interaction with Rad5. DNA Repair 2013, 12, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lin, A.; Zhou, C.; Blackwell, S.R.; Zhang, Y.; Wang, Z.; Feng, Q.; Guan, R.; Hanna, M.D.; Chen, Z.; et al. Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Sung, P.; Zhao, X. Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev. 2015, 29, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Kegel, A.; Sjogren, C. The Smc5/6 Complex: More Than Repair? Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Payne, F.; Colnaghi, R.; Rocha, N.; Seth, A.; Harris, J.; Carpenter, G.; Bottomley, W.E.; Wheeler, E.; Wong, S.; Saudek, V.; et al. Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J. Clin. Investig. 2014, 124, 4028–4038. [Google Scholar] [CrossRef] [PubMed]
- Van der Crabben, S.N.; Hennus, M.P.; McGregor, G.A.; Ritter, D.I.; Nagamani, S.C.S.; Wells, O.S.; Harakalova, M.; Chinn, I.K.; Alt, A.; Vondrova, L.; et al. Destabilized SMC5/6 complex leads to chromosome breakage syndrome with severe lung disease. J. Clin. Investig. 2016, 126, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Blobel, G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 4777–4782. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and Mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006, 127, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Machín, F.; Farmer, S.; Jarmuz, A.; Eydmann, T.; Dalgaard, J.Z.; Aragón, L. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 2005, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Agrawal, V.; Johnson, F.B. Homologous recombination-dependent rescue of deficiency in the structural maintenance of chromosomes (Smc) 5/6 complex. J. Biol. Chem. 2011, 286, 5119–5125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Choi, K.; Szakal, B.; Arenz, J.; Duan, X.; Ye, H.; Branzei, D.; Zhao, X. Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc. Natl. Acad. Sci. USA 2009, 106, 21252–21257. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Choi, K.; Bonner, J.; Chiba, T.; Kwon, Y.; Xu, Y.; Sanchez, H.; Wyman, C.; Niu, H.; Zhao, X.; et al. Restriction of replication fork regression activities by a conserved SMC complex. Mol. Cell 2014, 56, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Choi, K.; Bonner, J.N.; Szakal, B.; Chen, Y.-H.; Papusha, A.; Saro, D.; Niu, H.; Ira, G.; Branzei, D.; et al. Selective modulation of the functions of a conserved DNA motor by a histone fold complex. Genes Dev. 2015, 29, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Altmannova, V.; Luke-Glaser, S.; Henriksen, P.; Gallina, I.; Yang, X.; Choudhary, C.; Luke, B.; Krejci, L.; Lisby, M. Mte1 interacts with Mph1 and promotes crossover recombination and telomere maintenance. Genes Dev. 2016, 30, 700–717. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Papusha, A.; Choi, K.; Bonner, J.N.; Kumar, S.; Niu, H.; Kaur, H.; Zheng, X.-F.; Donnianni, R.A.; Lu, L.; et al. Differential regulation of the anti-crossover and replication fork regression activities of Mph1 by Mte1. Genes Dev. 2016, 30, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Yimit, A.; Kim, T.; Anand, R.P.; Meister, S.; Ou, J.; Haber, J.E.; Zhang, Z.; Brown, G.W. MTE1 functions with MPH1 in double-strand break repair. Genetics 2016, 203, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Dummer, A.M.; Su, Z.; Cherney, R.; Choi, K.; Denu, J.; Zhao, X.; Fox, C.A. Binding of the Fkh1 Forkhead Associated Domain to a Phosphopeptide within the Mph1 DNA Helicase Regulates Mating-Type Switching in Budding Yeast. PLoS Genet. 2016, 12, e1006094. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.E.; Ajazi, A.; Carotenuto, W.; Foiani, M.; Giannattasio, M. Rad53-mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep. 2015, 13, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 2005, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Jossen, R.; Colosio, A.; Frattini, C.; Carotenuto, W.; Cocito, A.; Doksani, Y.; Klein, H.; Gómez-González, B.; et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 2011, 146, 233–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Groden, J.; Orren, D.K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 2006, 45, 13939–13946. [Google Scholar] [CrossRef] [PubMed]
- Sarbajna, S.; West, S.C. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem. Sci. 2014, 39, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, W.; Liberi, G. Mitotic inter-homologue junctions accumulate at damaged DNA replication forks in recQ mutants. DNA Repair 2010, 9, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Zwicky, K.; Follonier, C.; Foiani, M.; Lopes, M.; Branzei, D. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; Mankouri, H.W.; Heidenblut, A.; McHugh, P.J.; Hickson, I.D. Pathways for Holliday junction processing during homologous recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 2011, 31, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Lopez, M.; Ceschia, A.; De Piccoli, G.; Colomina, N.; Pasero, P.; Aragon, L.; Torres-Rosell, J. The Smc5/6 complex is required for dissolution of DNA-mediated sister chromatid linkages. Nucleic Acids Res. 2010, 38, 6502–6512. [Google Scholar] [CrossRef] [PubMed]
- Jacome, A.; Gutierrez-Martinez, P.; Schiavoni, F.; Tenaglia, E.; Martinez, P.; Rodriguez-Acebes, S.; Lecona, E.; Murga, M.; Mendez, J.; Blasco, M.A.; et al. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 2015, 34, 2604–2619. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Lopez, M.; Villoria, M.T.; Esteras, M.; Jarmuz, A.; Torres-Rosell, J.; Clemente-Blanco, A.; Aragon, L. Sgs1’s roles in DNA end resection, HJ dissolution, and crossover suppression require a two-step SUMO regulation dependent on Smc5/6. Genes Dev. 2016, 30, 1339–1356. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.N.; Choi, K.; Xue, X.; Torres, N.P.; Szakal, B.; Wei, L.; Wan, B.; Arter, M.; Matos, J.; Sung, P.; et al. Smc5/6 mediated sumoylation of the Sgs1-Top3-Rmi1 complex promotes removal of recombination intermediates. Cell Rep. 2016, 16, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-López, M.; Pociño-Merino, I.; Sánchez, H.; Bueno, A.; Guasch, C.; Almedawar, S.; Bru-Virgili, S.; Garí, E.; Wyman, C.; Reverter, D.; et al. ATPase-Dependent Control of the Mms21 SUMO Ligase during DNA Repair. PLoS Biol. 2015, 13, e1002089. [Google Scholar] [CrossRef] [PubMed]
- Fasching, C.L.; Cejka, P.; Kowalczykowski, S.C.; Heyer, W.-D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell 2015, 57, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; de Muyt, A.; Lichten, M. Top3-Rmi1 DNA single-strand decatenase is integral to the formation and resolution of meiotic recombination intermediates. Mol. Cell 2015, 57, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wu, M.K.Y.; Zhang, R.; Hunter, N. Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Mol. Cell 2015, 57, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Glineburg, M.R.; Chavez, A.; Agrawal, V.; Brill, S.J.; Johnson, F.B. Resolution by Unassisted Top3 Points to Template Switch Recombination Intermediates during DNA Replication. J. Biol. Chem. 2013. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.G.; Matos, J. Hold your horSSEs: Controlling structure-selective endonucleases MUS81 and Yen1/GEN1. Front. Genet. 2015, 6, 253. [Google Scholar] [CrossRef] [PubMed]
- Gallo-Fernández, M.; Saugar, I.; Ortiz-Bazán, M.Á.; Vázquez, M.V.; Tercero, J.A. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 2012, 40, 8325–8335. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.S.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Cussiol, J.R.; Dibitetto, D.; Pellicioli, A.; Smolka, M.B. Slx4 scaffolding in homologous recombination and checkpoint control: Lessons from yeast. Chromosoma 2016. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Hang, L.E.; Zhao, X. Multi-BRCT scaffolds use distinct strategies to support genome maintenance. Cell Cycle 2016. [Google Scholar] [CrossRef]
- Chavdarova, M.; Marini, V.; Sisakova, A.; Sedlackova, H.; Vigasova, D.; Brill, S.J.; Lisby, M.; Krejci, L. Srs2 promotes Mus81-Mms4-mediated resolution of recombination intermediates. Nucleic Acids Res. 2015, 43, 3626–3642. [Google Scholar] [CrossRef] [PubMed]
- Marcomini, I.; Gasser, S.M. Nuclear organization in DNA end processing: Telomeres vs double-strand breaks. DNA Repair 2015, 32, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Beltrame, M.; Paonessa, G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science 1989, 243, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, S.; de Koning, L.; Gaillard, C.; Muselíková-Polanská, E.; Stros, M.; Strauss, F. Determinants of specific binding of HMGB1 protein to hemicatenated DNA loops. J. Mol. Biol. 2005, 353, 822–837. [Google Scholar] [CrossRef] [PubMed]
- Kamau, E.; Bauerle, K.T.; Grove, A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains. J. Biol. Chem. 2004, 279, 55234–55240. [Google Scholar] [CrossRef] [PubMed]
- Murugesapillai, D.; McCauley, M.J.; Huo, R.; Nelson Holte, M.H.; Stepanyants, A.; Maher, L.J.; Israeloff, N.E.; Williams, M.C. DNA bridging and looping by HMO1 provides a mechanism for stabilizing nucleosome-free chromatin. Nucleic Acids Res. 2014, 42, 8996–9004. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Xiao, L.; Grove, A. Yeast high mobility group protein HMO1 stabilizes chromatin and is evicted during repair of DNA double strand breaks. Nucleic Acids Res. 2015, 43, 5759–5770. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Huici, V.; Szakal, B.; Urulangodi, M.; Psakhye, I.; Castellucci, F.; Menolfi, D.; Rajakumara, E.; Fumasoni, M.; Bermejo, R.; Jentsch, S.; et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014, 33, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Tittel-Elmer, M.; Lengronne, A.; Davidson, M.B.; Bacal, J.; François, P.; Hohl, M.; Petrini, J.H.J.; Pasero, P.; Cobb, J.A. Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol. Cell 2012, 48, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Adachi, Y.; Yanagida, M. Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 2004, 430, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Mcaleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragón, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2013, 493, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Deursen, F.V.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2–7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Samora, C.P.; Saksouk, J.; Goswami, P.; Wade, B.O.; Singleton, M.R.; Bates, P.A.; Lengronne, A.; Costa, A.; Uhlmann, F. Ctf4 Links DNA Replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol. Cell 2016, 63, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Simon, A.C.; Ortiz Bazan, M.A.; Kilkenny, M.L.; Wirthensohn, D.; Wightman, M.; Matak-Vinkovíc, D.; Pellegrini, L.; Labib, K. Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol. Cell 2016, 63, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karras, G.I.; Fumasoni, M.; Sienski, G.; Vanoli, F.; Branzei, D.; Jentsch, S. Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol. Cell 2013, 49, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [PubMed]
- González-Prieto, R.; Muñoz-Cabello, A.M.; Cabello-Lobato, M.J.; Prado, F. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J. 2013, 32, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, D.O.; van Den Berg, J.; Medema, R.H. Breaks in the 45S rDNA lead to recombination-mediated loss of repeats. Cell Rep. 2016, 14, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is required to suppress deleterious resection of stressed replication forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. eLife 2013, 2, e00505. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Dusad, K.; Wright, W.D.; Grubb, J.; Budke, B.; Heyer, W.-D.; Connell, P.P.; Weichselbaum, R.R.; Bishop, D.K. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Res. 2015, 43, 3180–3196. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Zheng, X.; Epshtein, A.; Carey, J.N.; Bishop, D.K.; Klein, H.L. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Mol. Cell 2010, 39, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Flott, S.; Kwon, Y.; Pigli, Y.Z.; Rice, P.A.; Sung, P.; Jackson, S.P. Regulation of Rad51 function by phosphorylation. EMBO Rep. 2011, 12, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.-Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Waizenegger, A.; Barton, O.; Sürder, M.; Wright, W.D.; Heyer, W.-D.; Löbrich, M. Nek1 regulates Rad54 to orchestrate homologous recombination and replication fork stability. Mol. Cell 2016, 62, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Watanabe, K.; Wolanin, K.M.; Bartkova, J.; Wassing, I.E.; Watanabe, S.; Strauss, R.; Troelsgaard Pedersen, R.; Oestergaard, V.H.; Lisby, M.; et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 2016, 212, 281–288. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonner, J.N.; Zhao, X. Replication-Associated Recombinational Repair: Lessons from Budding Yeast. Genes 2016, 7, 48. https://doi.org/10.3390/genes7080048
Bonner JN, Zhao X. Replication-Associated Recombinational Repair: Lessons from Budding Yeast. Genes. 2016; 7(8):48. https://doi.org/10.3390/genes7080048
Chicago/Turabian StyleBonner, Jacob N., and Xiaolan Zhao. 2016. "Replication-Associated Recombinational Repair: Lessons from Budding Yeast" Genes 7, no. 8: 48. https://doi.org/10.3390/genes7080048
APA StyleBonner, J. N., & Zhao, X. (2016). Replication-Associated Recombinational Repair: Lessons from Budding Yeast. Genes, 7(8), 48. https://doi.org/10.3390/genes7080048