Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome


Abstract
:1. Introduction
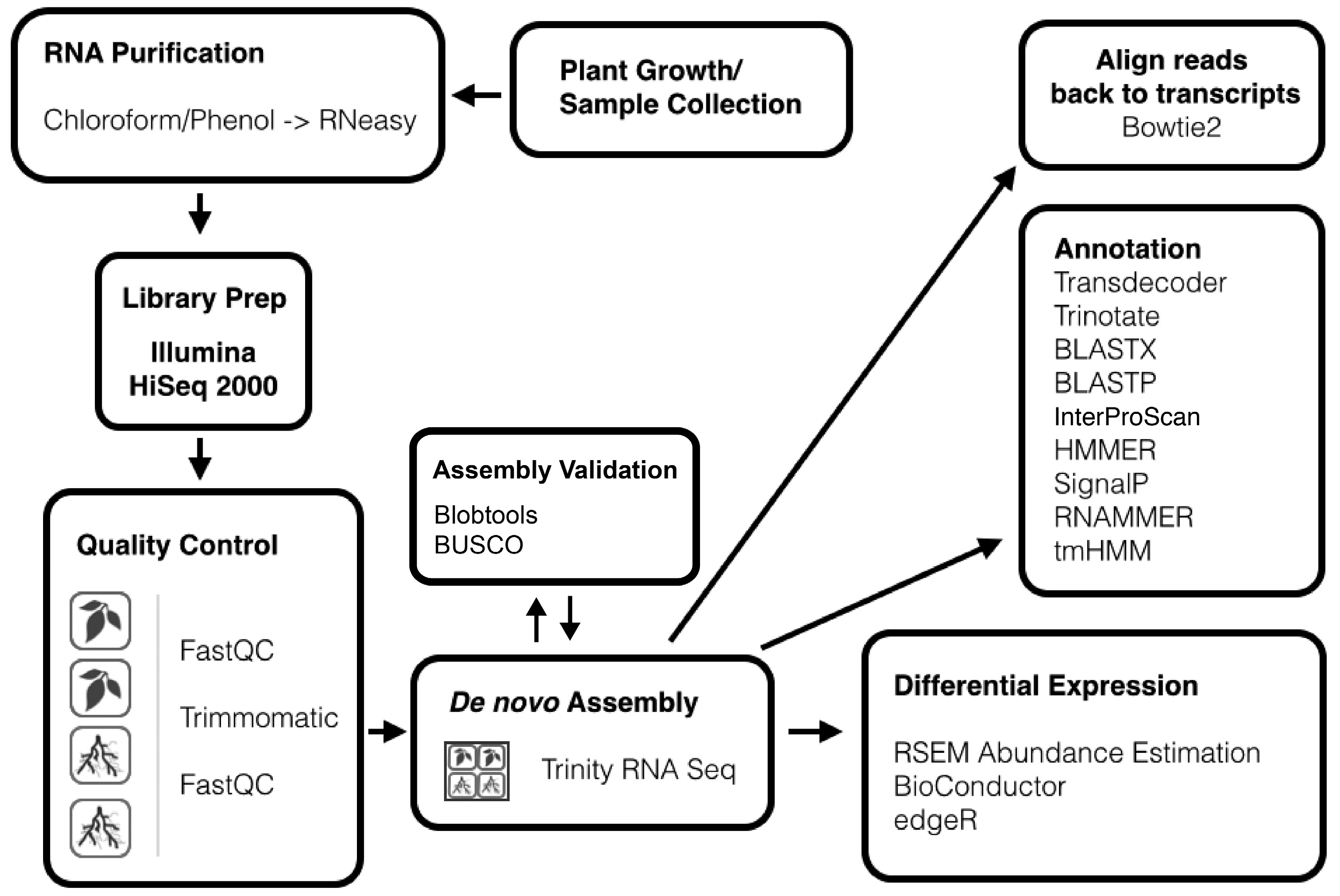
2. Materials and Methods
2.1. Axenic Cultured Seedlings and Plant Tissue
2.2. RNA Purification and Sequencing
2.3. De Novo Assembly of Poison Ivy Transcripts
2.4. Comparative Transcriptome Analysis of Anacardiaceae
3. Results and Discussion
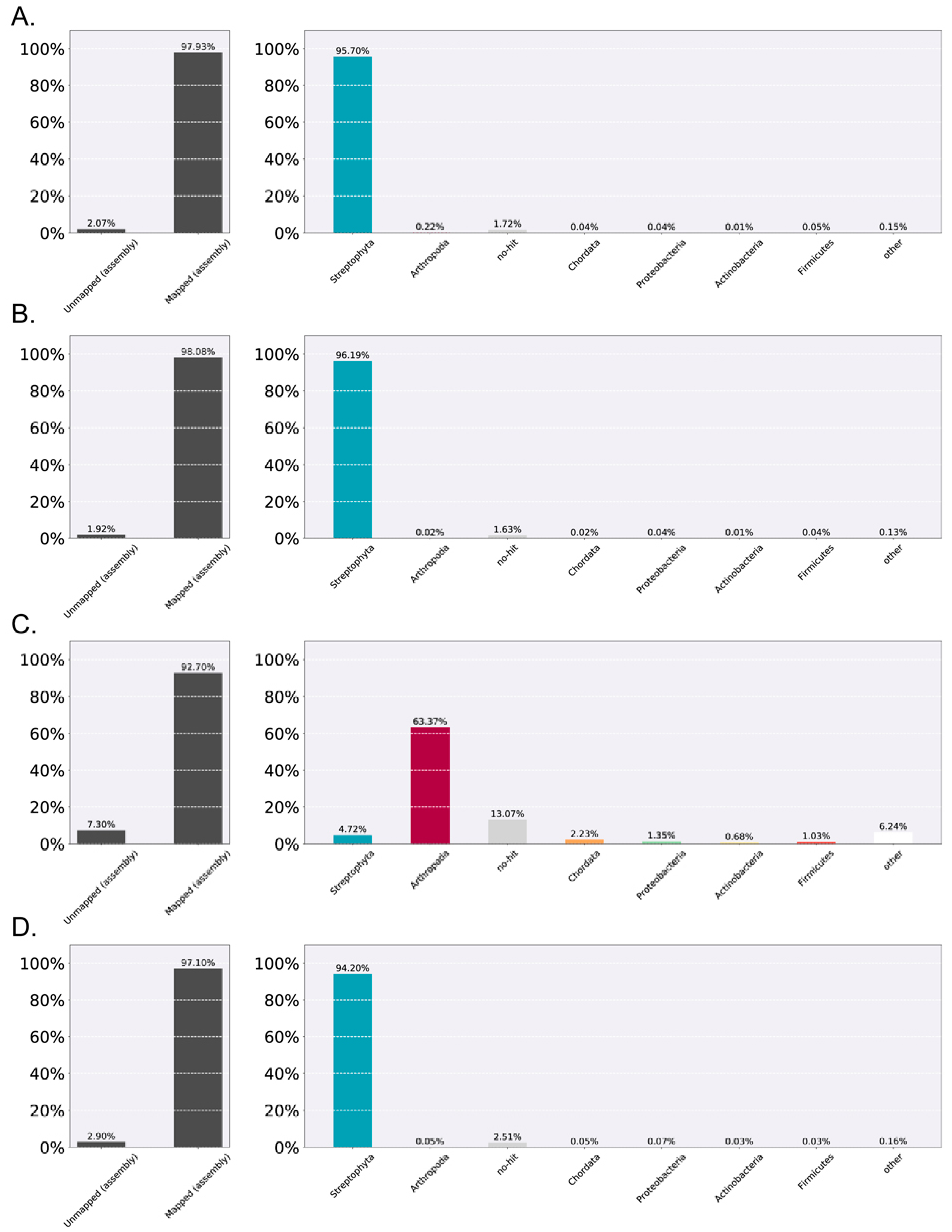
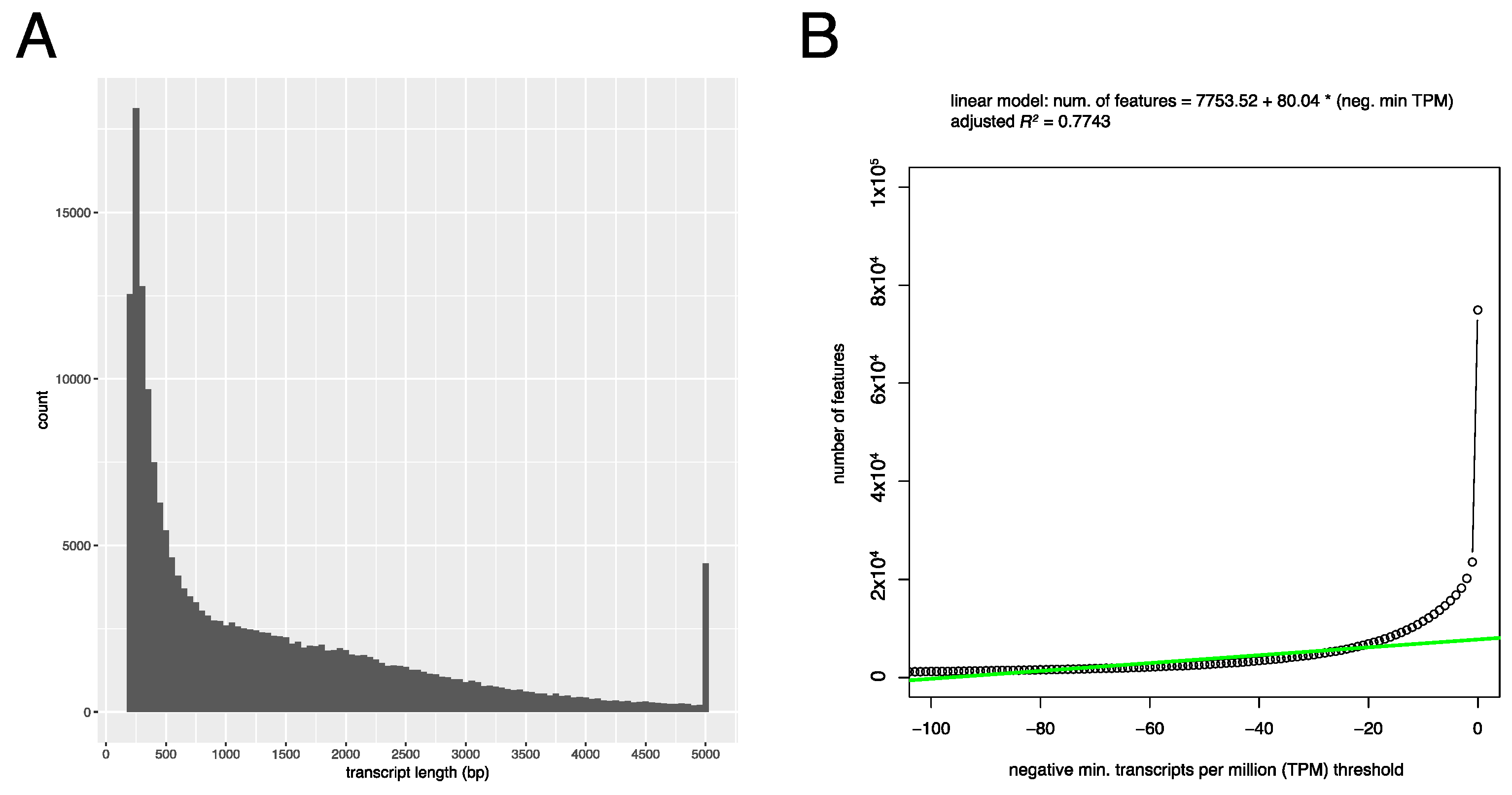
3.1. De Novo Assembly and Quality Control
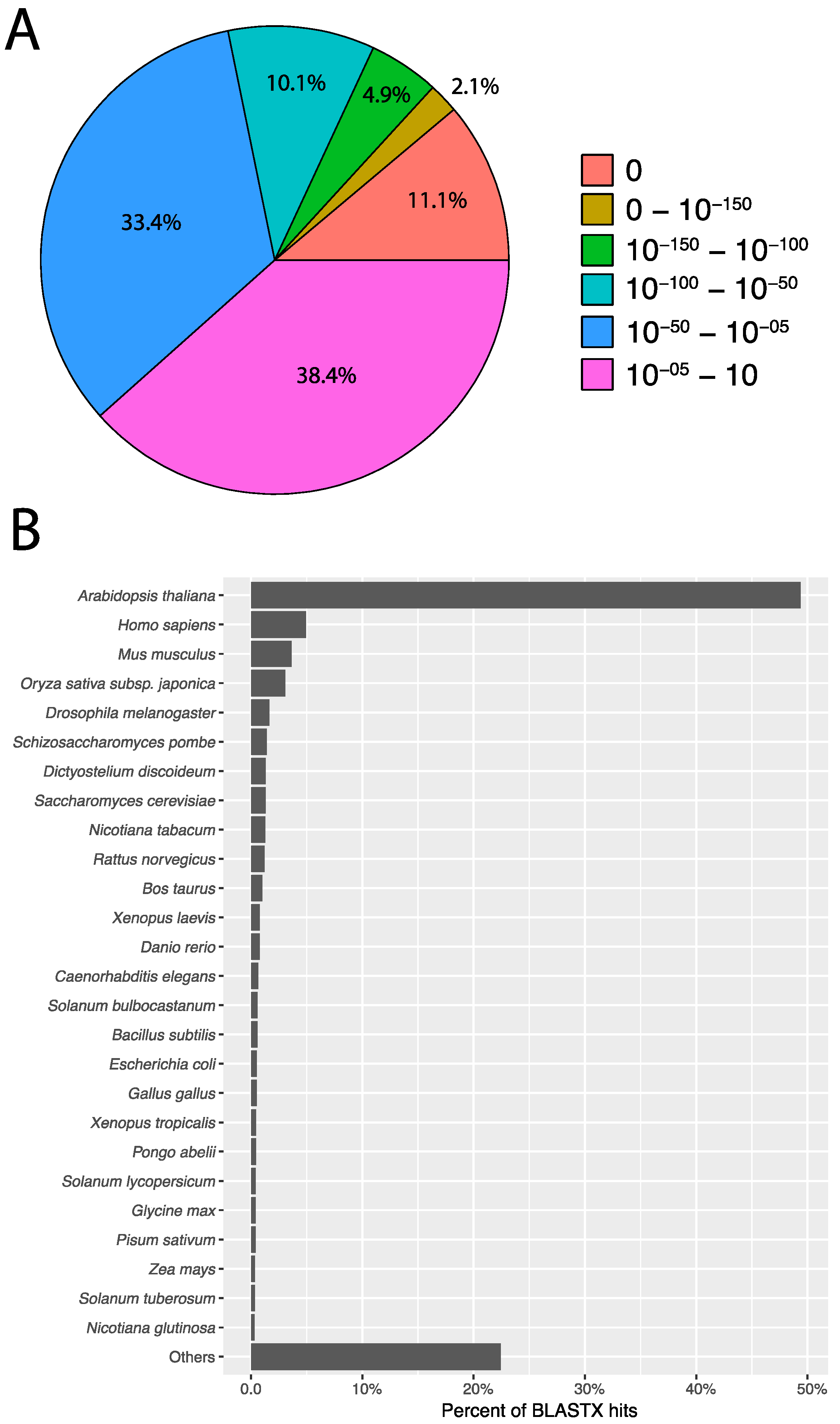
3.2. Transcriptome Annotation
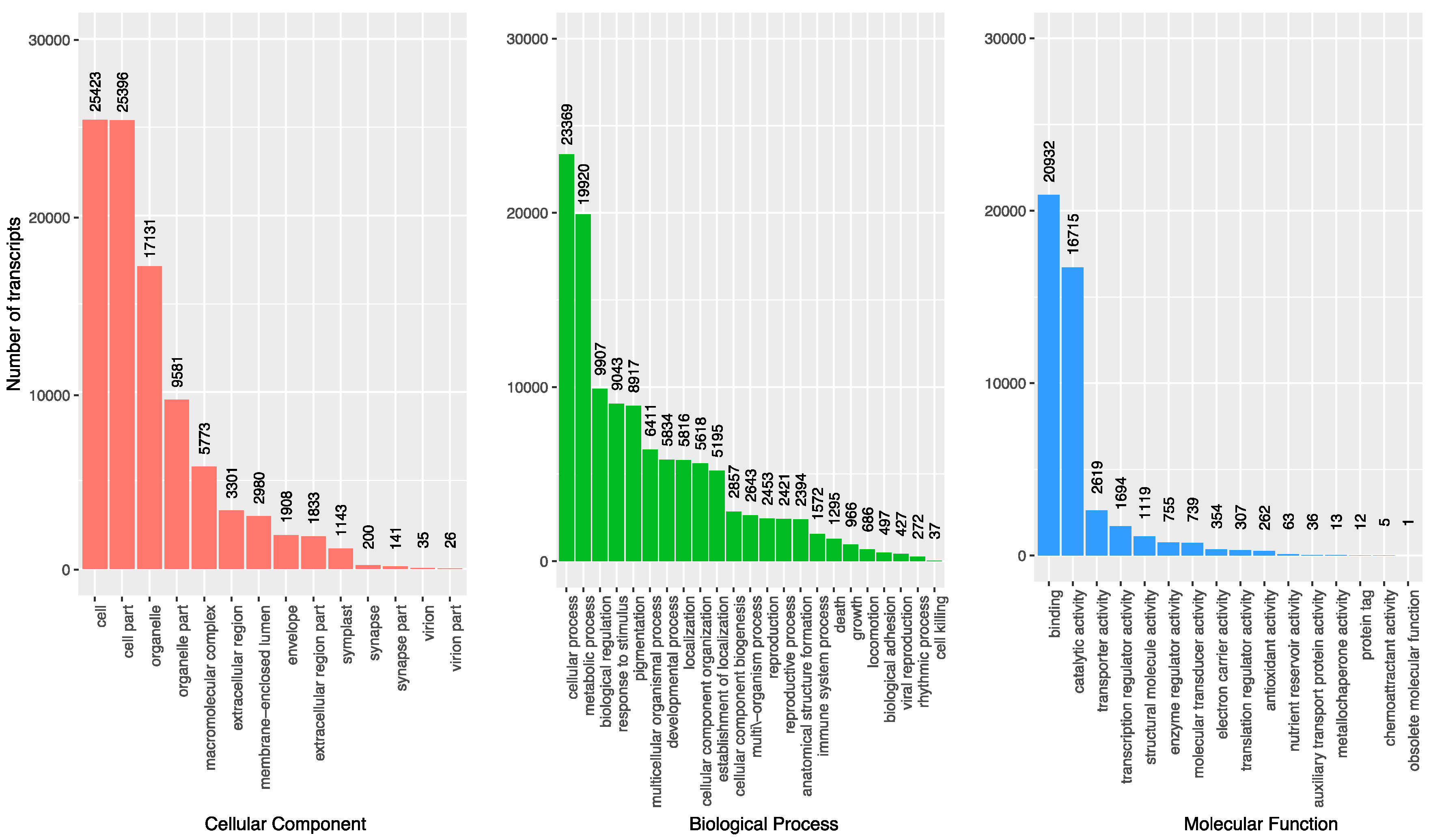
3.3. Gene Oontology Term Analysis
3.4. Conserved Protein Domain Analysis with InterProScan
3.5. Comparison of Leaf and Root Gene Expression
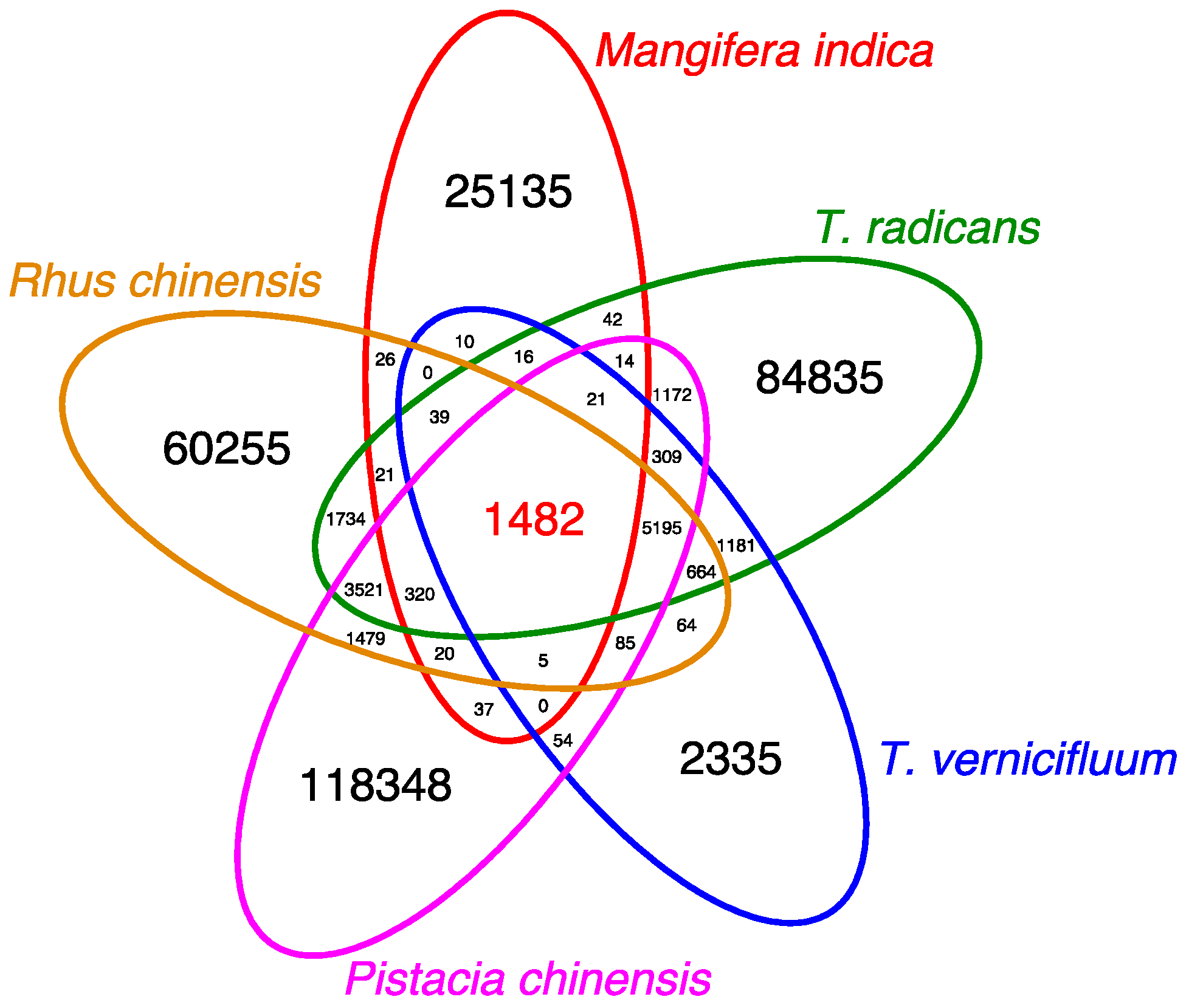
3.6. Pan-Anacardiaceae Transcriptome Comparison
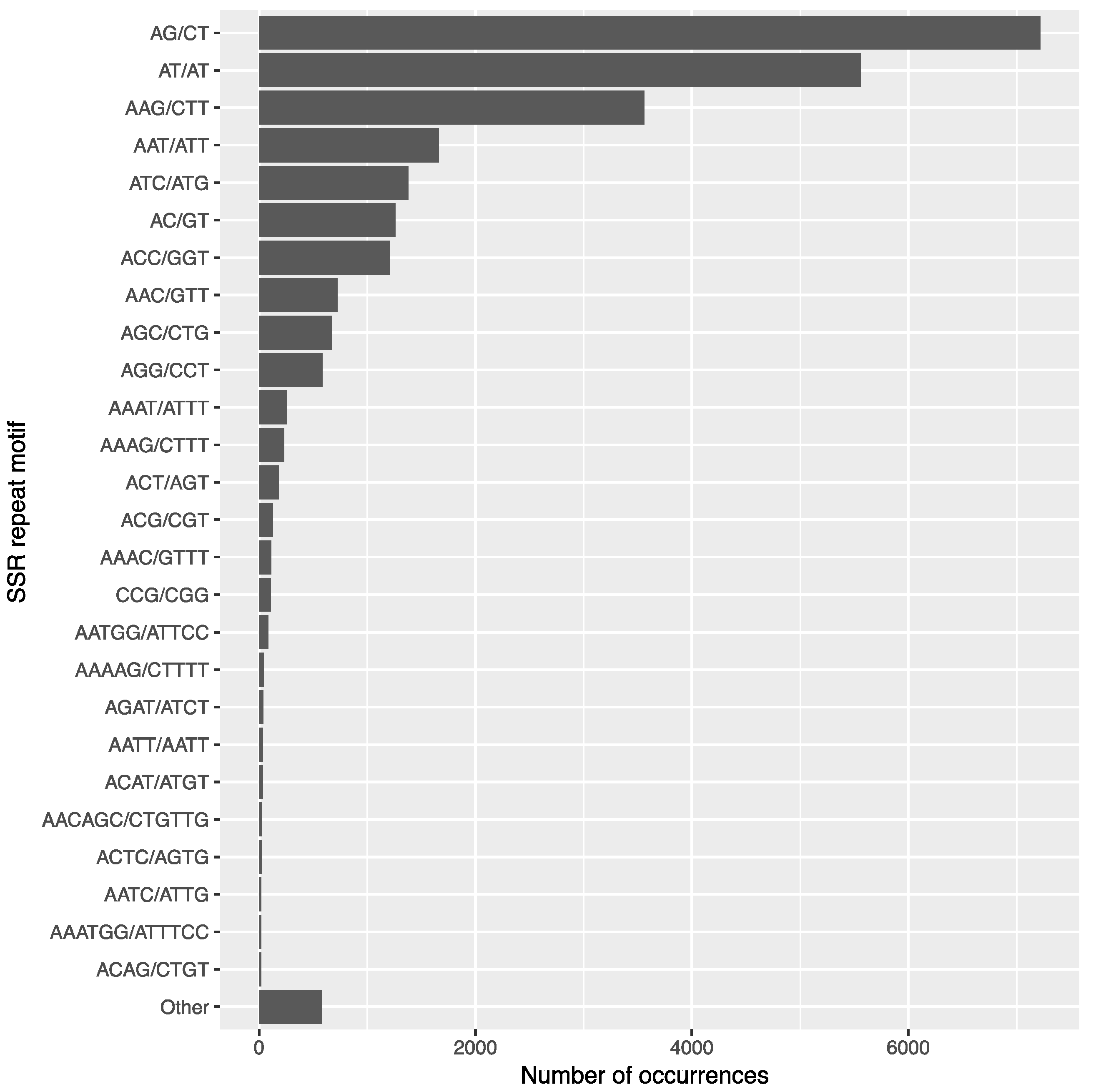
3.7. SSR Identification and Primer Design
4. Conclusions
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- Epstein, W.L. Plant-induced dermatitis. Ann. Emerg. Med. 1987, 16, 950–955. [Google Scholar] [CrossRef]
- Epstein, W.L. Occupational poison ivy and oak dermatitis. Derrmatol. Clin. 1994, 3, 511–516. [Google Scholar]
- Gayer, K.D.; Burnett, J.W. Toxicodendron dermatitis. Cutis 1988, 42, 99–100. [Google Scholar] [PubMed]
- Mitchell, J.; Rook, A. Botanical Dermatology: Plants and Plant Products Injurious to the Skin; Greengrass: Vancouver, BC, Canada, 1979; p. 787. [Google Scholar]
- Hill, G.A.; Mattacotti, V.; Graha, W.D. The toxic principle of the poison ivy. J. Am. Chem. Soc. 1934, 56, 2736–2738. [Google Scholar] [CrossRef]
- Majima, R. Uber den hauptbestandteil des japan-lacs, viii. Mitteilung: Stellung der doppelbindungen in der seitenkette des urushiols und beweisfuhrung, dab das urushiol eine mischung ist. Ber. Dtsch. Chem. Ges. 1922, 55, 172–191. (In German) [Google Scholar] [CrossRef]
- Majima, R.; Cho, S. Ueber einen hauptbestandteil des japanischen lackes. Ber. Dtsch. Chem. Ges. 1907, 40, 632. (In German) [Google Scholar] [CrossRef]
- Markiewitz, K.H.; Dawson, C.R. On the isolation of the allegenically active components of the toxic principle of poison ivy. J. Org. Chem. 1965, 30, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Symes, W.F.; Dawson, C.R. Separation and structural determination of the olefinic components of poison ivy urushiol, cardanol and cardol. Nature 1953, 171, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.C.; Waller, C.W.; Billets, S.; Elsohly, M.A. New GLC analysis of urushiol congeners in different plant parts of poison ivy, Toxicodendron radicans. J. Pharmacol. Sci. 1978, 67, 483–485. [Google Scholar] [CrossRef]
- Aziz, M.; Sturtevant, D.; Winston, J.; Collakova, E.; Jelesko, J.G.; Chapman, K.D. MALDI-MS imaging of urushiols in poison ivy stem. Molecules 2017, 22, 711. [Google Scholar] [CrossRef] [PubMed]
- Gillis, W.T. The systematics and ecology of poison-ivy and the poison-oaks (Toxicodendron, Anacardiaceae). Rhodora 1971, 73, 161–237. [Google Scholar]
- Penner, R.; Moodie, G.E.; Staniforth, R.J. The dispersal of fruits and seeds of poison-ivy, Toxicodendron radicans, by ruffed grouse, Bonasa umbellus, and squirrels, Tamiasciurus hudsonicus and Sciurus carolinensis. Can. Field-Nat. 1999, 113, 616–620. [Google Scholar]
- Talley, S.M.; Lawton, R.O.; Setzer, W.N. Host preferences of Rhus radicans (Anacardiaceae) in a southern deciduous harwood forest. Ecology 1996, 77, 1271–1276. [Google Scholar] [CrossRef]
- Mohan, J.E.; Ziska, L.H.; Schlesinger, W.H.; Thomas, R.B.; Sicher, R.C.; George, K.; Clark, J.S. Biomass and toxicity responses of poison ivy (Toxicodendron radicans) to elevated atmospheric CO2. Proc. Natl. Acad. Sci. USA 2006, 103, 9086–9089. [Google Scholar] [CrossRef] [PubMed]
- Ziska, L.H.; Sicher, R.C.; George, K.; Mohan, J.E. Rising atmospheric carbon dioxide and potential impacts on the growth and toxicity of poison ivy (Toxicodendron radicans). Weed Sci. 2007, 55, 288–292. [Google Scholar] [CrossRef]
- Gillis, W.T. Poison ivy and its kin. Arnoldia 1975, 35, 93–123. [Google Scholar]
- Allen, B.P.; Pauley, E.F.; Sharitz, R.R. Hurricane impacts on liana populations in an old-growth southeastern bottomland forest. J. Torrey Bot. Soc. 1997, 124, 34–42. [Google Scholar] [CrossRef]
- Allen, B.P.; Sharitz, R.R.; Goebel, P.C. Twelve years post-hurricane liana dynamics in an old-growth southeastern floodplain forest. For. Ecol. Manag. 2005, 218, 259–269. [Google Scholar] [CrossRef]
- Catling, P.M.; Sinclair, A.; Cuddy, D. Plant community composition and relationships of disturbed and undisturbed alvar woodland. Can. Field-Nat. 2002, 116, 571–579. [Google Scholar]
- Martin, A.C.; Zim, H.S.; Nelson, A.L. American Wildlife & Plants; Dover Publications, Inc.: New York, NY, USA, 1951; p. 500. [Google Scholar]
- Popay, I.; Field, R. Grazing animals as weed control agents. Weed Technol. 1996, 10, 217–231. [Google Scholar]
- Senchina, D.S. Fungal and animal associates of Toxicodendron spp. (Anacardiaceae) in North America. Perspect. Plant Ecol. Evol. Syst. 2008, 10, 197–216. [Google Scholar] [CrossRef]
- Suthers, H.B.; Bickal, J.M.; Rodewald, P.G. Use of successional habitat and fruit resources by songbirds during autumn migration in central New Jersey. Wilson Bull. 2000, 112, 249–260. [Google Scholar] [CrossRef]
- Senchina, D.S. Beetle interactions with poison ivy and poison oak (Toxicodendron p. Mill. Sect. Toxicodendron, Anacardiaceae). Coleopt. Bull. 2005, 59, 328–334. [Google Scholar] [CrossRef]
- Senchina, D.S.; Summerville, K.S. Great diversity of insect floral associates may partially explain ecological success of poison ivy (Toxicodendron radicans subps negundo greene gillis, Anacardiaceae). Gt. Lakes Entomol. 2007, 40, 120–128. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach; John Wiley & Sons: Chichester, UK, 1997; p. 466. [Google Scholar]
- Giessman, T.A. The biosynthesis of phenolic plant products. In The Biogenesis of Natural Compounds, 2nd ed.; Bernfeld, P., Ed.; Pergamon Press Ltd.: Oxford, UK, 1967; p. 1209. [Google Scholar]
- Jelesko, J.G.; Benhase, E.B.; Barney, J.N. Differential responses to light and nutrient availability by geographically isolated poison ivy accessions. Northeast. Nat. 2017, 24, 191–200. [Google Scholar] [CrossRef]
- Johnson, R.A.; Baer, H.; Kirkpatrick, C.H.; Dawson, C.R.; Khurana, R.G. Comparison of the contact allergenicity of the four pentadecylcatechols derived from poison ivy urushiol in human subjects. J. Allergy Clin. Immunol. 1972, 49, 27–35. [Google Scholar] [CrossRef]
- Benhase, E.B.; Jelesko, J.G. Germinating and culturing axenic poison ivy seedlings. HortScience 2013, 48, 1–5. [Google Scholar]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Ausubel, F.A.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/projects/fastqc (accessed on 26 April 2010).
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jones, M.; Koutsovoulos, G.; Clarke, M.; Blaxter, M. Blobology: Exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 2013, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.; Waterhouse, R.; Ioannidis, P.; Kriventseva, E.; Zdobnov, E. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.; Bowden, J.; Couger, M.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nat. Protocols 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Clements, J.; Eddy, S. Hmmer web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.; Eddy, S.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.; Brunak, S.; von, H.G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von, H.G.; Sonnhammer, E. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.; Staerfeldt, H.; Rognes, T.; Ussery, D. Rnammer: Consistent and rapid annotation of ribosomalRNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; McCarthy, D.; Smyth, G. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Wakefield, M.; Smyth, G.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Foster, Z.; Sharpton, T.; Grunwald, N. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comp. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.K.; Zhang, Y.; Chen, J.; Zhang, Z.J.; Wang, J.; Li, S.T.; Li, R.Q.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T. Misa—Microsatellite identification tool. Available online: http://pgrc.ipk-gatersleben.de/misa/ (accessed on 31 July 2017).
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B.; Cantalapiedra, C.; Garcia-Pereira, M.; Gordon, S.; Vogel, J.; Igartua, E.; Casas, A.; Vinuesa, P. Analysis of plant pan-genomes and transcriptomes with GET_HOMOLOGUES-EST, a clustering solution for sequences of the same species. Front. Plant Sci. 2017, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.H.; Girke, T. SystemPipeR: NGS workflow and report generation environment. BMC Bioinform. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.-L.; Sun, H.; Meng, Y.; Wen, J. Phylogenetic analysis of Toxicodendron (Anacardiaceae) and its biogeographic implications on the evolution of north temperate and tropical intercontinental disjunctions. J. Syst. Evol. 2009, 47, 416–430. [Google Scholar] [CrossRef]
- Lulin, H.; Xiao, Y.; Pei, S.; Wen, T.; Shangqin, H. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS ONE 2012, 7, e38653. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Ichihashi, Y.; Farhi, M.; Zumstein, K.; Townsley, B.; David-Schwartz, R.; Sinha, N. De novo assembly and characterization of the transcriptome of the parasitic weed dodder identifies genes associated with plant parasitism. Plant Physiol. 2014, 166, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ruhlman, T.; Mower, J.; Jansen, R. Comparative analyses of two Geraniaceae transcriptomes using next-generation sequencing. BMC Plant Biol. 2013, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Puniya, B.; Kulshreshtha, D.; Kumar, S.; Kaur, J.; Ramachandran, S.; Singh, K. De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PLoS ONE 2013, 8, e83336. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Wu, Y.; Chen, C.; Lei, J. De novo transcriptome assembly in chili pepper (Capsicum frutescens) to identify genes involved in the biosynthesis of capsaicinoids. PLoS ONE 2013, 8, e48156. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.; Zerbino, D.; Vingron, M.; Birney, E. Oases: Robust de novoRNA-Seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Kasson, M.T.; Pollok, J.R.; Benhase, E.B.; Jelesko, J.G. First report of seedling blight of eastern poison ivy (Toxicodendron radicans) by Colletotrichum fioriniae in Virginia. Plant Dis. 2014, 98, 995–996. [Google Scholar] [CrossRef]
- Marcelino, J.; Giordano, R.; Gouli, S.; Gouli, V.; Parker, B.L.; Skinner, M.; TeBeest, D.; Cesnik, R. Colletotrichum acutatum var. Fioriniae (teleomorph: Glomerella acutata var. Fioriniae var. Nov.) infection of a scale insect. Mycologia 2008, 100, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, J.A.; Gouli, S.; Parker, B.L.; Skinner, M.; Giordano, R. Entomopathogenic activity of a variety of the fungus, Colletotrichum acutatum, recovered from the elongate hemlock scale, Fiorinia externa. J. Insect Sci. 2009, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Ortigoza, C.J.; Sosa, V.; Aguilar-Ortigoza, M. Toxic phenols in various Anacardiaceae species. Econ. Bot. 2003, 57, 354–364. [Google Scholar] [CrossRef]
- Hsu, T.W.; Shih, H.C.; Ku, C.C.; Chiang, T.Y.; Chiang, Y.C. Characterization of 42 microsatellite markers from poison ivy, Toxicodendron radicans (Anacardiaceae). Int. J. Mol. Sci. 2013, 14, 20414–20426. [Google Scholar] [CrossRef] [PubMed]
- Barkley, F.A. A monographic study of Rhus and its immediate allies in North and Central America, including the West Indies. Ann. MO. Bot. Gard. 1937, 37, 265–498. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Leaves A | Leaves B | Roots A | Roots B | Total |
|---|---|---|---|---|---|
| Total # of paired reads | 97,494,163 | 93,384,797 | 30,270,702 | 128,496,278 | 349,645,940 |
| Total # of paired reads after trimming | 87,835,193 | 81,637,895 | 14,779,168 | 105,073,040 | 289,325,296 |
| % of paired reads surviving trimming | 90.09% | 87.42% | 48.82% | 81.77% | |
| GC content | 46% | 46.5% | 40% | 47% | |
| Total # of assembled contigs | 197,641 | ||||
| # of Trinity components | 74,937 | ||||
| Total assembled bases | 273,066,414 | ||||
| Mean contig length (bp) | 1381.63 | ||||
| Contig N50 (bp) | 2363 | ||||
| Contig GC% | 39.05 | ||||
| Longest contig (bp) | 16,102 | ||||
| # contigs > 1 kb | 93,444 | ||||
| Contigs with SwissProt BLASTX hits | 156,935 | ||||
| Contigs with KEGG annotation | 119,797 | ||||
| Contigs with GO annotation | 132,208 |
| Domain | Description | Transcripts with Domain |
|---|---|---|
| IPR027417 | P-loop containing nucleoside triphosphate hydrolase | 9503 |
| IPR000719 | Protein kinase domain | 7405 |
| IPR032675 | Leucine-rich repeat domain, L domain-like | 3946 |
| IPR011009 | Protein kinase-like domain | 2937 |
| IPR016040 | NAD(P)-binding domain | 2652 |
| IPR020683 | Ankyrin repeat-containing domain | 2482 |
| IPR011990 | Tetratricopeptide-like helical domain | 2273 |
| IPR029058 | Alpha/Beta hydrolase fold | 2022 |
| IPR000504 | RNA recognition motif domain | 1867 |
| IPR012677 | Nucleotide-binding alpha-beta plait domain | 1866 |
| MISA SSR Settings | Count |
| Total number of sequences examined | 197,641 |
| Total size of examined sequences (bp) | 273,066,414 |
| Total number of identified SSRs | 25,781 |
| Number of SSR containing sequences | 21,075 |
| Number of sequences containing more than 1 SSR | 3764 |
| Number of SSRs present in compound formation | 1843 |
| Unit Size | Number of SSRs |
| di-nucleotide | 14,044 |
| tri-nucleotide | 10,211 |
| quad-nucleotide | 835 |
| tetra-nucleotide | 316 |
| hexa-nucleotide | 375 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weisberg, A.J.; Kim, G.; Westwood, J.H.; Jelesko, J.G. Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome. Genes 2017, 8, 317. https://doi.org/10.3390/genes8110317
Weisberg AJ, Kim G, Westwood JH, Jelesko JG. Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome. Genes. 2017; 8(11):317. https://doi.org/10.3390/genes8110317
Chicago/Turabian StyleWeisberg, Alexandra J., Gunjune Kim, James H. Westwood, and John G. Jelesko. 2017. "Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome" Genes 8, no. 11: 317. https://doi.org/10.3390/genes8110317
APA StyleWeisberg, A. J., Kim, G., Westwood, J. H., & Jelesko, J. G. (2017). Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome. Genes, 8(11), 317. https://doi.org/10.3390/genes8110317