
Primary Cilia as a Possible Link between Left-Right Asymmetry and Neurodevelopmental Diseases


Abstract
:1. Introduction
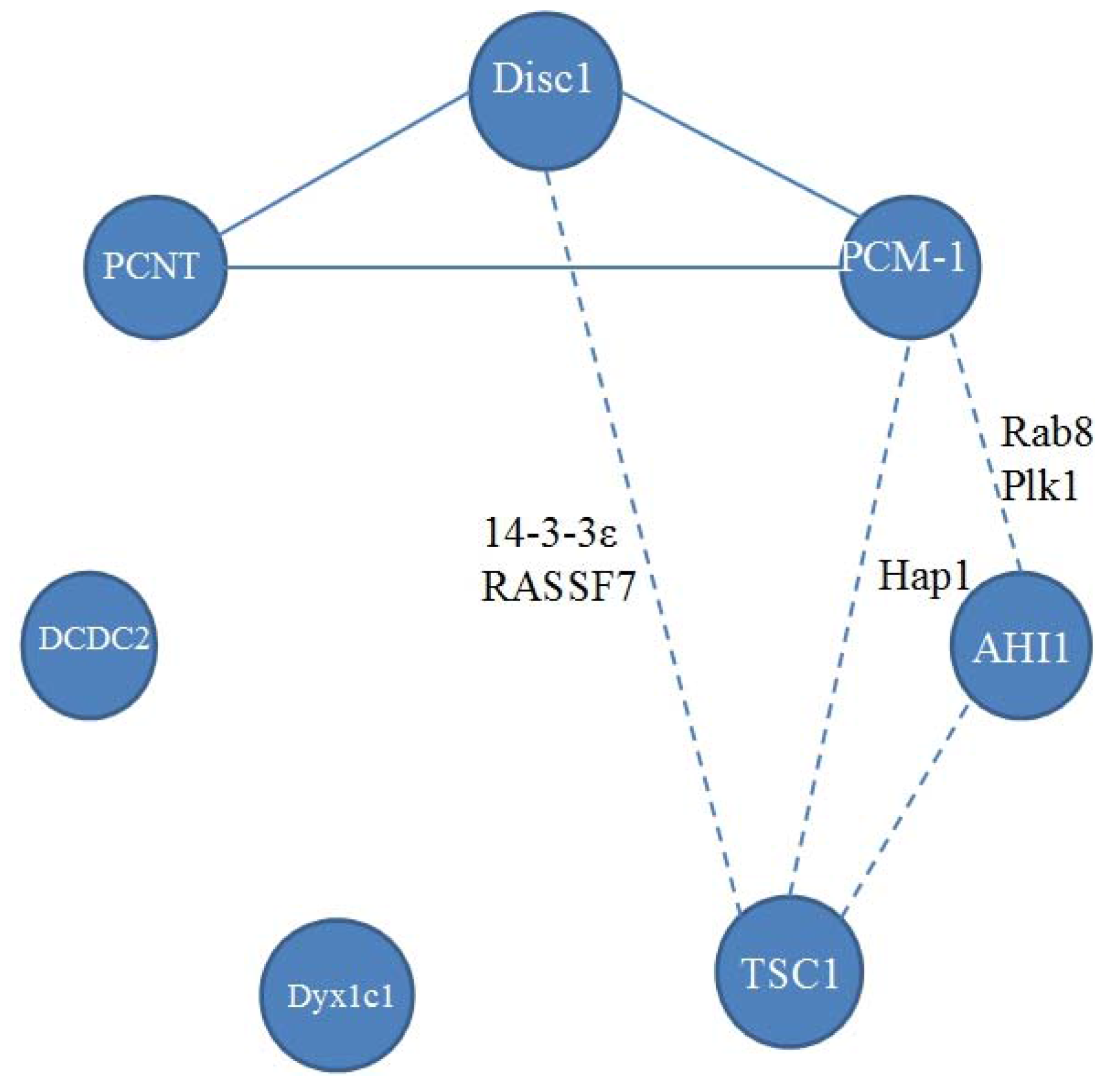
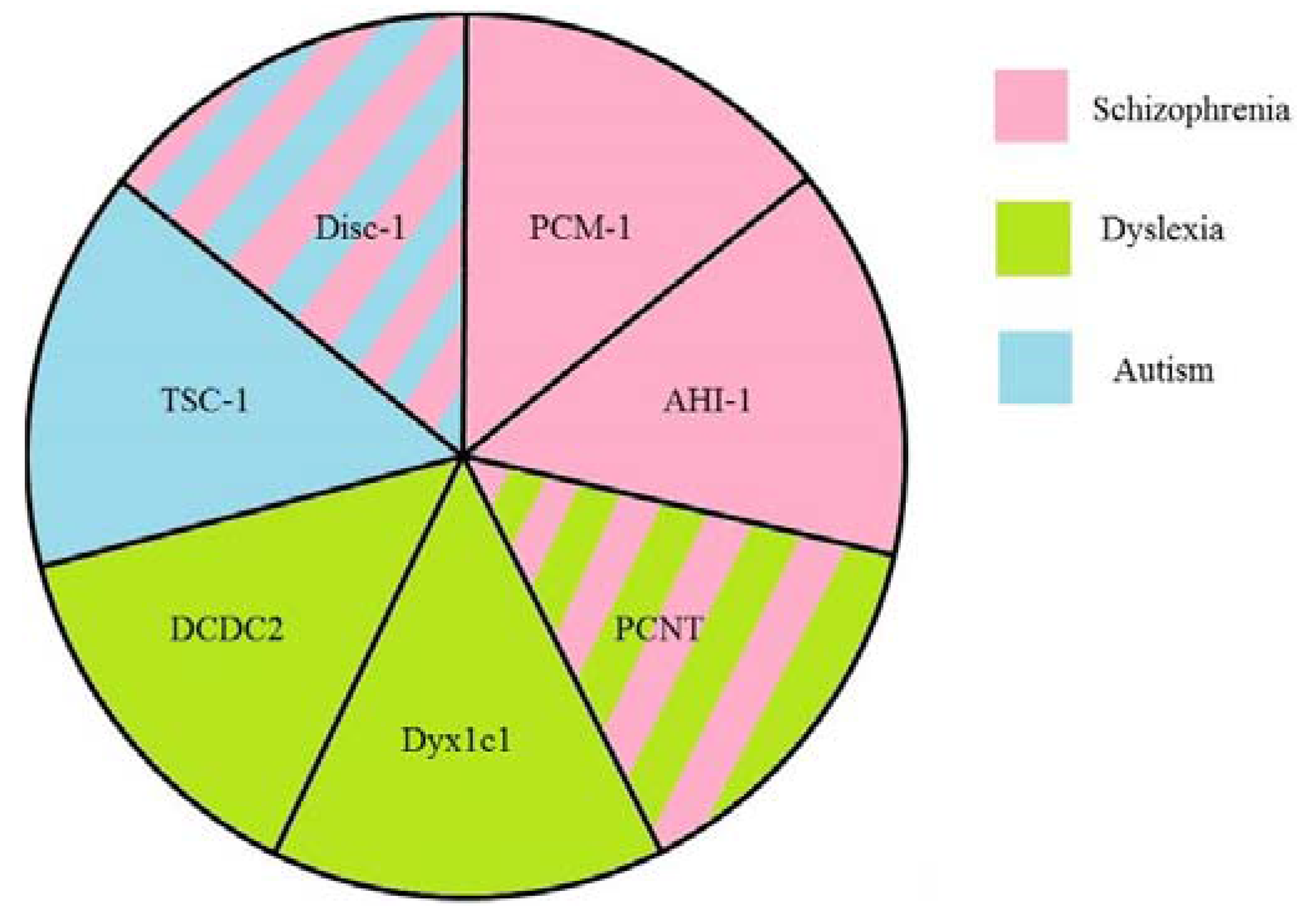
2. Disrupted in Schizophrenia 1 (Disc1)
3. Pericentriolar Material (PCM-1)
4. Pericentrin (PCNT)
5. Abelson Helper Integration Site 1 (AHI1)
6. Hamartin (Tuberous Sclerosis 1—TSC1)
7. DCDC2
8. DYX1C1
9. General Discussion
Acknowledgments
Conflicts of Interest
References
- Singla, V.; Reiter, J.F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Del Bigio, M.R. The ependyma: A protective barrier between brain and cerebrospinal fluid. Glia 1995, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Wichterle, H.; Gonzalez-Perez, O.; Cholfin, J.A.; Yamada, M.; Spassky, N.; Murcia, N.S.; Garcia-Verdugo, J.M.; Marin, O.; Rubenstein, J.L.; et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 2006, 311, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ibanez-Tallon, I.; Pagenstecher, A.; Fliegauf, M.; Olbrich, H.; Kispert, A.; Ketelsen, U.P.; North, A.; Heintz, N.; Omran, H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 2004, 13, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Thomson, P.A.; Malavasi, E.L.; Grunewald, E.; Soares, D.C.; Borkowska, M.; Millar, J.K. DISC1 genetics, biology and psychiatric illness. Front. Biol. 2013, 8, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Gleeson, J.G. Cilia in the nervous system: Linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 2011, 24, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.E.; Katsanis, N. The ciliopathies: A transitional model into systems biology of human genetic disease. Curr. Opin. Gen. Dev. 2012, 22, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Afzelius, B.A. A human syndrome caused by immotile cilia. Science 1976, 193, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Rott, H.D. Kartagener′s syndrome and the syndrome of immotile cilia. Hum. Genet. 1979, 46, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Shiratori, H.; Saijoh, Y.; Hamada, H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 2002, 418, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef]
- Kamura, K.; Kobayashi, D.; Uehara, Y.; Koshida, S.; Iijima, N.; Kudo, A.; Yokoyama, T.; Takeda, H. Pkd1l1 complexes with Pkd2 on motile cilia and functions to establish the left-right axis. Development 2011, 138, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Somlo, S.; Makova, S.; Tian, X.; Brueckner, M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 2003, 114, 61–73. [Google Scholar] [CrossRef]
- Babu, D.; Roy, S. Left-right asymmetry: cilia stir up new surprises in the node. Open Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Vogan, K.J.; Wagner, M.K.; Tabin, C.J.; Yost, H.J.; Brueckner, M. Conserved function for embryonic nodal cilia. Nature 2002, 418, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Amack, J.D.; Nyholm, M.K.; Harris, E.B.; Yost, H.J. Kupffer′s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 2005, 132, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Schweickert, A.; Weber, T.; Beyer, T.; Vick, P.; Bogusch, S.; Feistel, K.; Blum, M. Cilia-driven leftward flow determines laterality in Xenopus. Curr. Biol. 2007, 17, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Dathe, V.; Gamel, A.; Manner, J.; Brand-Saberi, B.; Christ, B. Morphological left-right asymmetry of Hensen’s node precedes the asymmetric expression of Shh. and Fgf-8 in the chick embryo. Anat. Embryol. 2002, 205, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Nishide, K.; Mugitani, M.; Kumano, G.; Nishida, H. Neurula rotation determines left-right asymmetry in ascidian tadpole larvae. Development 2012, 139, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Concha, M.L.; Burdine, R.D.; Russell, C.; Schier, A.F.; Wilson, S.W. A Nodal signaling pathway regulates the laterality of neuroanatomical asymmetries in the zebrafish forebrain. Neuron 2000, 28, 399–409. [Google Scholar] [CrossRef]
- Roussigne, M.; Bianco, I.H.; Wilson, S.W.; Blader, P. Nodal signaling imposes left-right asymmetry upon neurogenesis in the habenular nuclei. Development 2009, 136, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Barth, K.A.; Miklosi, A.; Watkins, J.; Bianco, I.H.; Wilson, S.W.; Andrew, R.J. Fsi. zebrafish show concordant reversal of laterality of viscera, neuroanatomy, and a subset of behavioral responses. Curr. Biol. 2005, 15, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Roussigne, M.; Blader, P.; Wilson, S.W. Breaking symmetry: The zebrafish as a model for understanding left-right asymmetry in the developing brain. Dev. Neurobiol. 2012, 72, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.N.; O′Craven, K.M.; Ticho, B.S.; Goldstein, A.M.; Makris, N.; Henson, J.W. Structural and functional brain asymmetries in human situs inversus totalis. Neurology 1999, 53, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- McManus, C. Reversed bodies, reversed brains, and (some) reversed behaviors: of zebrafish and men. Dev. Cell. 2005, 8, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Kanzaki, R.; Yoshibayashi, M.; Kamiya, T.; Sugishita, M. Dichotic listening in patients with situs inversus: Brain asymmetry and situs asymmetry. Neuropsychologia 1999, 37, 869–874. [Google Scholar] [CrossRef]
- Ihara, A.; Hirata, M.; Fujimaki, N.; Goto, T.; Umekawa, Y.; Fujita, N.; Terazono, Y.; Matani, A.; Wei, Q.; Yoshimine, T.; et al. Neuroimaging study on brain asymmetries in situs inversus totalis. J. Neurol. Sci. 2010, 288, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Malashichev, Y. Is there a link between visceral and neurobehavioral asymmetries in development and evolution? In Behavioral and Morphological Asymmetries in Veretbrates. Georgetown; Malashichev, Y.B., Deckel, W., Eds.; TX: Landes Bioscience, 2006; pp. 33–44. [Google Scholar]
- Malashichev, Y.B.; Wassersug, R.J. Left and right in the amphibian world: Which way to develop and where to turn? BioEssays 2004, 26, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Trulioff, A.S.; Malashichev, Y.B.; Ermakov, A.S. Artificial inversion of the left-right visceral asymmetry in vertebrates: Conceptual approaches and experimental solutions. Russ. J. Dev. Biol. 2015, 46, 307–325. [Google Scholar] [CrossRef]
- Clarke, J.M.; Zaidel, E. Anatomical-behavioral relationships: Corpus callosum morphometry and hemispheric specialization. Behav. Brain Res. 1994, 64, 185–202. [Google Scholar] [CrossRef]
- Witelson, S.F.; Nowakowski, R.S. Left out axons make men right: A hypothesis for the origin of handedness and functional asymmetry. Neuropsychologia 1991, 29, 327–333. [Google Scholar] [CrossRef]
- Brandler, W.M.; Morris, A.P.; Evans, D.M.; Scerri, T.S.; Kemp, J.P.; Timpson, N.J.; St Pourcain, B.; Smith, G.D.; Ring, S.M.; Stein, J.; et al. Common variants in left/right asymmetry genes and pathways are associated with relative hand skill. PLoS Genet. 2013, 9, e1003751. [Google Scholar] [CrossRef] [PubMed]
- Halpern, M.E.; Gunturkun, O.; Hopkins, W.D.; Rogers, L.J. Lateralization of the vertebrate brain: Taking the side of model systems. J. Neurosci. 2005, 25, 10351–10357. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.J. Asymmetry of brain and behavior in animals: Its development, function, and human relevance. Genesis 2014, 52, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.R.; Ziegler, D.A.; Deutsch, C.K.; O′Brien, L.M.; Kennedy, D.N.; Filipek, P.A.; et al. Brain asymmetries in autism and developmental language disorder: A nested whole-brain analysis. Brain 2005, 128, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Robichon, F.; Levrier, O.; Farnarier, P.; Habib, M. Developmental dyslexia: Atypical cortical asymmetries and functional significance. Eur. J. Neurol. 2000, 7, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Pujol, J.; Cardoner, N.; Benlloch, L.; Urretavizcaya, M.; Deus, J.; Losilla, J.M.; Bakardjiev, A.I.; Hodgson, J.; Takeoka, M.; Makris, N.; et al. CSF spaces of the Sylvian fissure region in severe melancholic depression. Neuroimage 2002, 15, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Crow, T.J.; Done, D.J.; Sacker, A. Cerebral lateralization is delayed in children who later develop schizophrenia. Schizophr. Res. 1996, 22, 181–185. [Google Scholar] [CrossRef]
- Petty, R.G. Structural asymmetries of the human brain and their disturbance in schizophrenia. Schizophr. Bull. 1999, 25, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, B.A. Mental symptoms occurring in Kartagener′s syndrome. Am. J. Psychiatry 1962, 118, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Glick, I.D.; Graubert, D.N. Kartagener′s syndrome and schizophrenia: A report of a case with chromosomal studies. Am. J. Psychiatry 1964, 121, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Mohan, I.; Lowe, M.; Sundram, S. Comorbid situs inversus totalis and schizophrenia in a young male. Aust. N. Z. J. Psychiatry 2013, 47, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Quast, T.M.; Sippert, J.D.; Sauve, W.M.; Deutsch, S.I. Comorbid presentation of Kartagener′s syndrome and schizophrenia: Support of an etiologic hypothesis of anomalous development of cerebral asymmetry? Schizophr. Res. 2005, 74, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Ermiş, A.; Turkcan, A.; Ceylan, M.E.; Maner, A.F. Kartagener Sendromu ve Psikotik Bozukluk: Olgu Sunumu. J. Psychiatry Neurol. Sci. 2009, 22, 32–35. (In Turkish) [Google Scholar]
- Kondziella, D.; Lycke, J. Autism spectrum disorders: does cilia dysfunction in embryogenesis play a role? Acta Neuropsychiatr. 2008, 20, 227–228. [Google Scholar] [CrossRef]
- Suzuki, T.; Washio, Y.; Aritaki, M.; Fujinami, Y.; Shimizu, D.; Uji, S.; Hashimoto, H. Metamorphic pitx2 expression in the left habenula correlated with lateralization of eye-sidedness in flounder. Dev. Growth Differ. 2009, 51, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Compagnucci, C.; Fish, J.; Depew, M.J. Left-right asymmetry of the gnathostome skull: Its evolutionary, developmental, and functional aspects. Genesis 2014, 52, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.; von Zastrow, M. DISC1 regulates primary cilia that display specific dopamine receptors. PLoS ONE 2010, 5, e10902. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.A.; Kandpal, G.; Ma, L.; Austin, C.P. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: Regulation and loss of interaction with mutation. Hum. Mol. Genet. 2003, 12, 1591–1608. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling and psychiatric disease: Bipolar disorder and schizophrenia. Cell Tissue Res. 2014, 357, 477–492. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Adams, R.R.; Christie, S.; Buchanan, S.R.; Porteous, D.J.; Millar, J.K. Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol. Cell. Neurosci. 2004, 26, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.U.; Jeong, J.; Lee, H.; Mun, J.Y.; Kim, J.H.; Lee, J.S.; Nguyen, M.D.; Han, S.S.; Suh, P.G.; Park, S.K. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc. Natl. Acad. Sci. USA 2010, 107, 17785–17790. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Coyle, J.T. Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 2011, 35, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Tan, P.L.; Kubo, K.I.; Engelhard, C.; Ishizuka, K.; Kubo, A.; Tsukita, S.; Pulver, A.E.; Nakajima, K.; Cascella, N.G.; et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: A candidate for psychiatric illnesses. Arch. Gen. Psychiatry 2008, 65, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, Q.; Zhang, X.; Zhang, B.; Zhuo, X.; Liu, J.; Jiang, Q.; Zhang, C. PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. J. Cell Sci. 2013, 126, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Yun, B.A.; Kittappa, R.; Anderegg, A.M.; Chang, W.W.; Taketo, M.M.; McKay, R.D.; Awatramani, R.B. Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nature Neurosci. 2009, 12, 125–131. [Google Scholar] [CrossRef]
- Doxsey, S.J.; Stein, P.; Evans, L.; Calarco, P.D.; Kirschner, M. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 1994, 76, 639–650. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamagiwa, A.; Nishimura, T.; Mukai, H.; Ono, Y. Centrosomal proteins CG-NAP and kendrin provide microtubule nucleation sites by anchoring γ-tubulin ring complex. Mol. Biol. Cell 2002, 13, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Karkar, K.M.; May, S.R.; Cobos, I.; Thwin, M.T.; Long, J.E.; Ashique, A.M.; Zarbalis, K.; Rubenstein, J.L.; Peterson, A.S. A mutation in the pericentrin gene causes abnormal interneuron migration to the olfactory bulb in mice. Dev. Biol. 2010, 340, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Chih, B.; Liu, P.; Chinn, Y.; Chalouni, C.; Komuves, L.G.; Hass, P.E.; Sandoval, W.; Peterson, A.S. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nature Cell Biol. 2012, 14, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-C.; Tong, Z.J.; Westfall, J.E.; Ault, J.G.; Page-McCaw, P.S.; Ferland, R.J. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 2009, 18, 3926–3941. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Hynes, A.M.; Eley, L.; Inglis, D.; Chaudhry, B.; Dawe, H.R.; Sayer, J.A. Modelling a ciliopathy: Ahi1 knockdown in model systems reveals an essential role in brain, retinal, and renal development. Cell. Mol. Life Sci. 2012, 69, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, S.; Pae, C.-U.; Han, C.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Balzarro, B.; Alberti, S.; De Ronchi, D.; Serretti, A. Abelson helper integration site-1 gene variants on major depressive disorder and bipolar disorder. Psychiatry Invest. 2014, 11, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Torri, F.; Akelai, A.; Lupoli, S.; Sironi, M.; Amann-Zalcenstein, D.; Fumagalli, M.; Dal Fiume, C.; Ben-Asher, E.; Kanyas, K.; Cagliani, R.; et al. Fine mapping of AHI1 as a schizophrenia susceptibility gene: From association to evolutionary evidence. FASEB J. 2010, 24, 3066–3082. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1—Independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Meikle, L.; Pollizzi, K.; Egnor, A.; Kramvis, I.; Lane, H.; Sahin, M.; Kwiatkowski, D.J. Response of a neuronal model of tuberous sclerosis to mTOR inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 2008, 28, 5422–5432. [Google Scholar] [CrossRef] [PubMed]
- DiBella, L.M.; Park, A.; Sun, Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 2009, 18, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Hengstschlager, M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 2008, 658, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Massinen, S.; Hokkanen, M.-E.; Matsson, H.; Tammimies, K.; Tapia-Paez, I.; Dahlstrom-Heuser, V.; Kuja-Panula, J.; Burghoorn, J.; Jeppsson, K.E.; Swoboda, P.; et al. Increased expression of the dyslexia candidate gene DCDC2 affects length and signaling of primary cilia in neurons. PLoS ONE 2011, 6, e20580. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, G.; Vesterlund, L.; Hultenby, K.; Tapia-Paez, I.; Kere, J. The Zebrafish Orthologue of the Dyslexia Candidate Gene DYX1C1 Is Essential for Cilia Growth and Function. PLoS ONE 2013, 8, e63123. [Google Scholar] [CrossRef] [PubMed]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nature Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paramasivam, M.; Thomas, A.; Bai, J.; Kaminen-Ahola, N.; Kere, J.; Voskuil, J.; Rosen, G.D.; Galaburda, A.M.; Loturco, J.J. DYX1C1 functions in neuronal migration in developing neocortex. Neuroscience 2006, 143, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.D.; Bai, J.; Wang, Y.; Fiondella, C.G.; Threlkeld, S.W.; LoTurco, J.J.; Galaburda, A.M. Disruption of neuronal migration by RNAi of Dyx1c1 results in neocortical and hippocampal malformations. Cereb. Cortex 2007, 17, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- St Clair, D.; Blackwood, D.; Muir, W.; Walker, M.; Carothers, A.; Spowart, G.; Gosden, C.; Evans, H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990, 336, 13–16. [Google Scholar] [CrossRef]
- Millar, J.K.; Wilson-Annan, J.C.; Anderson, S.; Christie, S.; Taylor, M.S.; Semple, C.A.; Devon, R.S.; St Clair, D.M.; Muir, W.J.; Blackwood, D.H.; et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genetics 2000, 9, 1415–1423. [Google Scholar] [CrossRef]
- Kilpinen, H.; Ylisaukko-Oja, T.; Hennah, W.; Palo, O.M.; Varilo, T.; Vanhala, R.; Nieminen-von Wendt, T.; Von Wendt, L.; Paunio, T.; Peltonen, L. Association of DISC1 with autism and Asperger syndrome. Mol. Psychiatry 2008, 13, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, R.; Numakawa, T.; Ohnishi, T.; Kumamaru, E.; Yagasaki, Y.; Ishimoto, T.; Mori, T.; Nemoto, K.; Adachi, N.; Izumi, A.; et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum. Mol. Genet. 2006, 15, 3024–3033. [Google Scholar] [CrossRef] [PubMed]
- Thomson, P.A.; Macintyre, D.J.; Hamilton, G.; Dominiczak, A.; Smith, B.H.; Morris, A.; Evans, K.L.; Porteous, D.J. Association of DISC1 variants with age of onset in a population-based sample of recurrent major depression. Mol. Psychiatry 2013, 18, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.A.; Goldman, D.; Jaeger, J.; Persaud, S.; Kane, J.M.; Lipsky, R.H.; Malhotra, A.K. Disrupted in schizophrenia 1 (DISC1): Association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am. J. Hum. Genet. 2004, 75, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Hennah, W.; Thomson, P.; McQuillin, A.; Bass, N.; Loukola, A.; Anjorin, A.; Blackwood, D.; Curtis, D.; Deary, I.J.; Harris, S.E.; et al. DISC1 association, heterogeneity and interplay in schizophrenia and bipolar disorder. Mol. Psychiatry 2009, 14, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Nwulia, E.; Chang, J.; Balkissoon, R.; Ishizuka, K.; Chen, H.; Zandi, P.; McInnis, M.G.; Sawa, A. Differential expression of disrupted-in-schizophrenia (DISC1) in bipolar disorder. Biol. Psychiatry 2006, 60, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Inglis, P.N.; Leitch, C.C.; Efimenko, E.; Zaghloul, N.A.; Mok, C.A.; Davis, E.E.; Bialas, N.J.; Healey, M.P.; Héon, E.; et al. An essential role for DYF-11/MIP-T3 in assembling functional intraflagellar transport complexes. PLoS Genet. 2008, 4, e1000044. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Brandon, N.J. Regulation of the cytoskeleton by Disrupted-in-Schizophrenia 1 (DISC1). Mol. Cell. Neurosci. 2011, 48, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, H.R.; Gleeson, J.G. The centrosome in neuronal development. Trends Neurosci. 2007, 30, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Kam, A.; Kubo, K.I.; Tomoda, T.; Takaki, M.; Youn, R.; Ozeki, Y.; Sawamura, N.; Park, U.; Kudo, C.; Okawa, M.; et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005, 7, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Tomoda, T.; Chang, J.; Takaki, M.; Zhan, C.; Morita, M.; Cascio, M.B.; Elashvili, S.; Koizumi, H.; Takanezawa, Y.; et al. DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum. Mol. Genet. 2006, 15, 3313–3323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Charych, E.I.; Pulito, V.L.; Lee, J.B.; Graziane, N.M.; Crozier, R.A.; Revilla-Sanchez, R.; Kelly, M.P.; Dunlop, A.J.; Murdoch, H.; et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol. Psychiatry 2011, 16, 1006–1023. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K.; et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 2009, 136, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Randall, A.D.; Kurihara, M.; Brandon, N.J.; Brown, J.T. Disrupted in schizophrenia 1 and synaptic function in the mammalian central nervous system. Eur. J. Neurosci. 2014, 39, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Duan, X.; Liu, C.Y.; Jang, M.H.; Guo, J.U.; Pow-anpongkul, N.; Kang, E.; Song, H.; Ming, G.L. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 2009, 63, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, H.; Mackie, S.; Collins, D.M.; Hill, E.V.; Bolger, G.B.; Klussmann, E.; Porteous, D.J.; Millar, J.K.; Houslay, M.D. Isoformselective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J. Neurosci. 2007, 27, 9513–9524. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Liu, C.Y.; Zhang, F.; Duan, X.; Wen, Z.; Song, J.; Feighery, E.; Lu, B.; Rujescu, D.; St Clair, D.; et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 2012, 148, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Graziane, N.M.; Gu, Z.; Yan, Z. DISC1 protein regulates γ-aminobutyric acid, type A (GABAA) receptor trafficking and inhibitory synaptic transmission in cortical neurons. J. Biol. Chem. 2015, 290, 27680–27687. [Google Scholar] [PubMed]
- Levchenko, A.; Davtian, S.; Freylichman, O.; Zagrivnaya, M.; Kostareva, A.; Malashichev, Y. Betacatenin in schizophrenia: Possibly deleterious novel mutation. Psychiatry Res. 2015, 228, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Shen, N.; Jin, H.; Liu, D.; Miao, X.; Zhu, L.Q. GSK-3beta polymorphism discriminates bipolar disorder and schizophrenia: A systematic meta-analysis. Mol. Neurobiol. 2013, 48, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Tucci, V.; Kleefstra, T.; Hardy, A.; Heise, I.; Maggi, S.; Willemsen, M.H.; Hilton, H.; Esapa, C.; Simon, M.; Buenavista, M.T.; et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Invest. 2014, 124, 1468–1482. [Google Scholar] [CrossRef] [PubMed]
- Balczon, R.; Bao, L.; Zimmer, W.E. PCM-1, A 228-kD centrosome autoantigen with a distinct cell cycle distribution. J. Cell Biol. 1994, 124, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Sasaki, H.; Yuba-Kubo, A.; Tsukita, S.; Shiina, N. Centriolar satellites: Molecular characterization, Atp-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 1999, 147, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Tsukita, S. Non-membranous granular organelle consisting of PCM-1: Subcellular distribution and cell-cycle-dependent assembly/disassembly. J. Cell Sci. 2003, 116, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Vladar, E.K.; Stearns, T. Molecular characterization of centriole assembly in ciliated epithelial cells. J. Cell Biol. 2007, 178, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Stowe, T.R.; Wilkinson, C.J.; Iqbal, A.; Stearns, T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol. Biol. Cell 2012, 23, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Krishnaswami, S.R.; Gleeson, J.G. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 2008, 17, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Keryer, G.; Pineda, J.R.; Liot, G.; Kim, J.; Dietrich, P.; Benstaali, C.; Smith, K.; Cordelières, F.P.; Spassky, N.; Ferrante, R.J.; et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 2011, 121, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.A.; Prosser, S.L.; Romio, L.; Hirst, R.A.; O′Callaghan, C.; Woolf, A.S.; Fry, A.M. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 2011, 124, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, S.L.; Hodgkinson, C.A.; Harrison, P.J. DISC-1 Leu607Phe alleles differentially affect centrosomal PCM1 localization and neurotransmitter release. Mol. Psychiatry 2009, 14, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, S.L.; Walker, M.; Hyde, T.M.; Kleinman, J.E.; Harrison, P.J. The DISC1 Ser704Cys substitution affects centrosomal localisation of its binding partner PCM1 in glia in human brain. Hum. Mol. Genet. 2010, 19, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Gurling, H.M.; Critchley, H.; Datta, S.R.; McQuillin, A.; Blaveri, E.; Thirumalai, S.; Pimm, J.; Krasucki, R.; Kalsi, G.; Quested, D.; et al. Genetic association and brain morphology studies and the chromosome 8p22 pericentriolar material 1 (PCM1) gene in susceptibility to schizophrenia. Arch. Gen. Psychiatry. 2006, 63, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; McQuillin, A.; Rizig, M.; Blaveri, E.; Thirumalai, S.; Kalsi, G.; Lawrence, J.; Bass, N.J.; Puri, V.; Choudhury, K.; et al. A threonine to isoleucine missense mutation in the pericentriolar material 1 gene is strongly associated with schizophrenia. Mol. Psychiatry 2010, 15, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, R.; Ohi, K.; Yasuda, Y.; Fukumoto, M.; Yamamori, H.; Kamino, K.; Morihara, T.; Iwase, M.; Kazui, H.; Numata, S.; et al. No association between the PCM1 gene and schizophrenia: A multi-center case-control study and a meta-analysis. Schizophr. Res. 2011, 129, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Takaki, M.; Okahisa, Y.; Mizuki, Y.; Kodama, M.; Ujike, H.; Uchitomi, Y. Four polymorphisms of the pericentriolar material 1 (PCM1) gene are not associated with schizophrenia in a Japanese population. Psychiatry Res. 2014, 216, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Zoubovsky, S.; Oh, E.C.; Cash-Padgett, T.; Johnson, A.W.; Hou, Z.; Mori, S.; Gallagher, M.; Katsanis, N.; Sawa, A.; Jaaro-Peled, H.; et al. Neuroanatomical and behavioral deficits in mice haploinsufficient for Pericentriolar material 1 (Pcm1). Neurosci. Res. 2015, 98, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Jurczyk, A.; Gromley, A.; Redick, S.; Agustin, J.S.; Witman, G.; Pazour, G.J.; Peters, D.J.; Doxsey, S. Pericentrin forms a complex with intraflagellar transport proteins and polycystin-2 and is required for primary cilia assembly. J. Cell Biol. 2004, 166, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Yamada, K.; Iwayama, Y.; Toyota, T.; Takei, N.; Iwata, Y.; Suzuki, K.; Sekine, Y.; Matsuzaki, H.; et al. Gene and expression analyses reveal enhanced expression of pericentrin 2 (PCNT2) in bipolar disorder. Biol. Psychiatry 2008, 63, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Yamada, K.; Iwayama, Y.; Toyota, T.; Takei, N.; et al. Association studies and gene expression analyses of the DISC1-interacting molecules, pericentrin 2 (PCNT2) and DISC1—Binding zinc finger protein (DBZ), with schizophrenia and with bipolar disorder. Am. J. Med. Genet. 2009, 150, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Numata, S.; Nakataki, M.; Iga, J.I.; Tanahashi, T.; Nakadoi, Y.; Ohi, K.; Iwata, Y.; Suzuki, K.; Sekine, Y.; Matsuzaki, H.; et al. Association study between the pericentrin (PCNT) gene and schizophrenia. Neuromol. Med. 2010, 12, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Katayama, T.; Tohyama, M.; Ogawa, N. DISC1 localizes to the centrosome by binding to kendrin. Biochem. Biophys. Res. Commun. 2004, 317, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Poelmans, G.; Engelen, J.J.M.; Lent-Albrechts, V.; Smeets, H.J.; Schoenmakers, E.; Franke, B.; Buitelaar, J.K.; Wuisman-Frerker, M.; Erens, W.; Steyaert, J.; et al. Identification of novel dyslexia candidate genes through the analysis of a chromosomal deletion. Am. J. Med. Genet. 2009, 150, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Ferland, R.J.; Eyaid, W.; Collura, R.V.; Tully, L.D.; Hill, R.S.; Al-Nouri, D.; Al-Rumayyan, A.; Topcu, M.; Gascon, G.; Bodell, A.; et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nature Genet. 2004, 36, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Dixon-Salazar, T.; Silhavy, J.L.; Marsh, S.E.; Louie, C.M.; Scott, L.C.; Gururaj, A.; Al-Gazali, L.; Al-Tawari, A.A.; Kayserili, H.; Sztriha, L.; et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004, 75, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Louie, C.M.; Caridi, G.; Lopes, V.S.; Brancati, F.; Kispert, A.; Lancaster, M.A.; Schlossman, A.M.; Otto, E.A.; Leitges, M.; Gröne, H.J.; et al. AHI1 is required for outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nature Genet. 2010, 42, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Gleeson, J.G. The role of primary cilia in neuronal function. Neurobiol. Dis. 2010, 38, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Schroth, J.; Gleeson, J.G. Subcellular spatial regulation of canonical Wnt signaling at the primary cilium. Nature Cell Biol. 2011, 13, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Xu, X.; Lin, Y.-F.; Wang, C.-E.; Rong, J.; Cheng, D.; Peng, J.; Jiang, X.; Li, S.H.; Li, X.J. Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Invest. 2008, 118, 2785–2795. [Google Scholar] [CrossRef] [PubMed]
- Eley, L.; Gabrielides, C.; Adams, M.; Johnson, C.A.; Hildebrandt, F.; Sayer, J.A. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008, 74, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Han, S.B.; Choi, B.I.; Lee, D.; Kee, S.H.; Kim, H.S.; Sun, W.; Kim, H. Regulation of AHI1 expression in adult rat brain: Implication in hypothalamic feeding control. Biochem. Biophys. Res. Commun. 2009, 390, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Juric-Sekhar, G.; Adkins, J.; Doherty, D.; Hevner, R.F. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012, 123, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Amann-Zalcenstein, D.; Avidan, N.; Kanyas, K.; Ebstein, R.P.; Kohn, Y.; Hamdan, A.; Ben-Asher, E.; Karni, O.; Mujaheed, M.; Segman, R.H.; et al. AHI1, a pivotal neurodevelopmental gene, and C6orf217 are associated with susceptibility to schizophrenia. Eur. J. Hum. Genet. 2006, 14, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.; Giegling, I.; Cichon, S.; Hansen, T.; Rasmussen, H.B.; Nielsen, J.; Jürgens, G.; Muglia, P.; Hartmann, A.M.; Strengman, E.; et al. A large replication study and meta-analysis in European samples provides further support for association of AHI1 markers with schizophrenia. Hum. Mol. Genet. 2010, 19, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.; Sigmundsson, T.; Steinberg, S.; Sigurdsson, E.; Haraldsson, M.; Magnusdottir, B.B.; Frigge, M.L.; Kong, A.; Gulcher, J.; Thorsteinsdottir, U.; et al. Support for involvement of the AHI1 locus in schizophrenia. Eur. J. Hum. Genet. 2007, 15, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Rivero, O.; Reif, A.; Sanjuan, J.; Molto, M.D.; Kittel-Schneider, S.; Najera, C.; Toepner, T.; Lesch, K.P. Impact of the AHI1 gene on the vulnerability to schizophrenia: A case-control association study. PLoS ONE 2010, 5, e12254. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, S.; Pae, C.-U.; Han, C.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Balzarro, B.; Alberti, S.; De Ronchi, D.; Serretti, A.; et al. The influence of AHI1 variants on the diagnosis and treatment outcome in schizophrenia. Int. J. Mol. Sci. 2015, 16, 2517–2529. [Google Scholar] [CrossRef] [PubMed]
- Slonimsky, A.; Levy, I.; Kohn, Y.; Rigbi, A.; Ben-Asher, E.; Lancet, D.; Agam, G.; Lerer, B. Lymphoblast and brain expression of AHI1 and the novel primate-specific gene, C6orf217, in schizophrenia and bipolar disorder. Schizophr. Res. 2010, 120, 159–166. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; Van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, G.; Li, S.; Brown, S.J.; Braverman, R.; Vass, W.C.; Cheadle, J.P.; Halley, D.J.; Sampson, J.R.; Wienecke, R.; DeClue, J.E. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000, 19, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.J.; Manning, B.D. Tuberous sclerosis: A GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 2005, 14, R251–R258. [Google Scholar] [CrossRef] [PubMed]
- Astrinidis, A.; Senapedis, W.; Henske, E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.S.; Aldred, M.; von Ruhland, C.; Harris, R.; Sandford, R.; Cheadle, J.P. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum. Mol. Genet. 2009, 18, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.-H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; de Vries, P.J.; Silva, A.J. From mTOR to cognition: Molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J. Intellect. Disabil. Res. 2009, 53, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.R. Phenotypes of the tuberous sclerosis complex with a revision of diagnostic criteria. Ann. N. Y. Acad. Sci. 1991, 615, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wataya-Kaneda, M. Mammalian target of rapamycin and tuberous sclerosis complex. J. Dermatol. Sci. 2015, 79, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Mak, B.C.; Takemaru, K.I.; Kenerson, H.L.; Moon, R.T.; Yeung, R.S. The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J. Biol. Chem. 2003, 278, 5947–5951. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol. Autism 2012. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Smith, S.D.; Hager, K.; Held, M.; Liu, J.; Olson, R.K.; Pennington, B.F.; DeFries, J.C.; Gelernter, J.; O′Reilly-Pol, T.; et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc. Natl. Acad. Sci. USA 2005, 102, 17053–17058. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Anthoni, H.; Dahdouh, F.; Konig, I.R.; Hillmer, A.M.; Kluck, N.; Manthey, M.; Plume, E.; Warnke, A.; Remschmidt, H.; et al. Strong genetic evidence of DCDC2 as a susceptibility gene for dyslexia. Am. J. Hum. Genet. 2006, 78, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 Mutations Cause a Renal-Hepatic Ciliopathy by Disrupting Wnt Signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Grati, M.H.; Chakchouk, I.; Ma, Q.; Bensaid, M.; Desmidt, A.; Turki, N.; Yan, D.; Baanannou, A.; Mittal, R.; Driss, N.; et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum. Mol. Genet. 2015, 24, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Marino, C.; Meng, H.; Mascheretti, S.; Rusconi, M.; Cope, N.; Giorda, R.; Molteni, M.; Gruen, J.R. DCDC2 genetic variants and susceptibility to developmental dyslexia. Psychiatric Genetics 2012, 22, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Paracchini, S.; Scerri, T.; Dennis, M.; Cope, N.; Hill, G.; Moskvina, V.; Walter, J.; Richardson, A.J.; Owen, M.J.; et al. Further evidence that the KIAA0319 gene confers susceptibility to developmental dyslexia. Mol. Psychiatry 2006, 11, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Jamadar, S.; Powers, N.R.; Meda, S.A.; Gelernter, J.; Gruen, J.R.; Pearlson, G.D. Genetic influences of cortical grey matter in language-related regions in healthy controls and schizophrenia. Schizophrenia Res. 2011, 129, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Darki, F.; Peyrard-Janvid, M.; Matsson, H.; Kere, J.; Klingberg, T. Three dyslexia susceptibility genes, DYX1C1, DCDC2, and KIAA0319, affect temporo-parietal white matter structure. Biol. Psychiatry 2012, 72, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef] [PubMed]
- Ivliev, A.E.; AC′t Hoen, P.A.C.; van Roon-Mom, W.M.; Peters, D.J.; Sergeeva, M.G. Exploring the transcriptome of ciliated cells using in silico dissection of human tissues. PLoS ONE 2012, 7, e35618. [Google Scholar] [CrossRef] [PubMed]
- Hoh, R.A.; Stowe, T.R.; Turk, E.; Stearns, T. Transcriptional rogram of ciliated epithelial cells reveals new cilium and centrosome components and links to human disease. PLoS ONE 2012, 7, e52166. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Ho, C.S.; Chou, C.H.; Waye, M.M. Association of the rs3743205 variant of DYX1C1 with dyslexia in Chinese children. Behav. Brain Funct. 2011. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, S.; Ang, Q.W.; Stanley, F.J.; Monaco, A.P.; Pennell, C.E.; Whitehouse, A.J.O. Analysis of dyslexia candidate genes in the Raine cohort representing the general Australian population. Genes Brain Behav. 2011, 10, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.K.; Siddaiah, A.; Padakannaya, P.; Ramachandra, N.B. Association of SNPs of DYX1C1 with developmental dyslexia in an Indian population. Psychiatric Genet. 2014, 24, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Rendall, A.R.; Tarkar, A.; Contreras-Mora, H.M.; LoTurco, J.J.; Fitch, R.H. Deficits in learning and memory in mice with a mutation of the candidate dyslexia susceptibility gene Dyx1c1. Brain Lang. 2015. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.P.; Grimes, D.T. Mouse models of ciliopathies: The state of the art. Dis. Model. Mech. 2012, 5, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J. The neuropathology of schizophrenia. Brain 1999, 122, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Tabares-Seisdedos, R.; Escamez, T.; Martinez-Gimenez, J.A.; Balanza, V.; Salazar, J.; Selva, G.; Rubio, C.; Vieta, E.; Geijo-Barrientos, E.; Martinez-Aran, A.; et al. Variations in genes regulating neuronal migration predict reduced prefrontal cognition in schizophrenia and bipolar subjects from mediterranean Spain: A preliminary study. Neuroscience 2006, 139, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Galaburda, A.M.; Sherman, G.F.; Rosen, G.D.; Abolitz, F.; Geschwind, N. Developmental dyslexia: Four consecutive patients with cortical anomalies. J. Neurol. 1985, 18, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.L.; Cotter, D.R.; Everall, I.P. Density and distribution of white matter neurons in schizophrenia, bipolar disorder and major depressive disorder: No evidence for abnormalities of neuronal migration. Mol. Psychiatry 2002, 7, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.; Hoogenraad, C.C. Centrosomes, microtubules and neuronal development. Mol. Cell. Neurosci. 2011, 48, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Delaval, B.; Doxsey, S.J. Pericentrin in cellular function and disease. J. Cell Biol. 2010, 188, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Guadiana, S.M.; Semple-Rowland, S.; Daroszewski, D.; Madorsky, I.; Breunig, J.J.; Mykytyn, K.; Sarkisian, M.R. Arborization of dendrites by developing neocortical neurons is dependent on primary cilia and type 3 adenylyl cyclase. J. Neurosci. 2013, 33, 2626–2638. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia-1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nature Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Margolis, R.L.; Reading, S.A.; Pletnikov, M.; Coyle, J.T. Neurobiology of schizophrenia. Neuron 2006, 52, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Hasan, A.; Gruber, O.; Falkai, P. Schizophrenia as a disorder of disconnectivity. Eur. Archives Psychiatry Clin. Neurosci. 2011, 261, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Hardan, A.; di Nemi, S.U.; Perez, J.; Soares, J.C.; Barale, F. Brain anatomy and development in autism: Review of structural MRI studies. Brain Res. Bull. 2003, 61, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Cawthon, B.; Clinkscales, W.; Bruce, A.; Winzenburger, P.; Ess, K.C. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb. Cortex 2012, 22, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARS to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Currier, T.A.; Etchegaray, M.A.; Haight, J.L.; Galaburda, A.M.; Rosen, G.D. The effects of embryonic knockdown of the candidate dyslexia susceptibility gene homologue Dyx1c1 on the distribution of GABAergic neurons in the cerebral cortex. Neurosci. Biobehav. Rev. 2011, 172, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Pradhan, D.A.; Ming, G.L.; Song, H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends Neurosci. 2007, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Petryshen, T.L.; Middleton, F.A.; Tahl, A.R.; Rockwell, G.N.; Purcell, S.; Aldinger, K.A.; Kirby, A.; Morley, C.P.; McGann, L.; Gentile, K.L.; et al. Genetic investigation of chromosome 5q GABAA receptor subunit genes in schizophrenia. Mol. Psychiatry 2005, 10, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Volk, D.W.; Hashimoto, T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: A novel target for the treatment of working memory dysfunction. Psychopharmacology 2004, 174, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Berretta, S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27. [Google Scholar] [CrossRef]
- Coghlan, S.; Horder, J.; Inkster, B.; Mendez, M.A.; Murphy, D.G.; Nutt, D.J. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 2012, 36, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Breunig, J.J.; Sarkisian, M.R.; Arellano, J.I.; Morozov, Y.M.; Ayoub, A.E.; Sojitra, S.; Wang, B.; Flavell, R.A.; Rakic, P.; Town, T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Colamarino, S.A.; Song, H.J.; Desire, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signaling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Freyberg, Z.; Ferrando, S.J.; Javitch, J.A. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 2009, 167, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14–3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.L.; Anderlid, B.M.; Lindstrand, A.; van Ravenswaaij-Arts, C.; Ganesamoorthy, D.; Lundin, J.; Martin, C.L.; Douglas, J.; Nowak, C.; Adam, M.P.; et al. Further molecular and clinical delineation of co-locating 17p13. 3 microdeletions and microduplications that show distinctive phenotypes. J. Med. Genet. 2010, 47, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Capra, V.; Mirabelli-Badenier, M.; Stagnaro, M.; Rossi, A.; Tassano, E.; Gimelli, S.; Gimelli, G. Identification of a rare 17p13.3 duplication including the BHLHA9 and YWHAE genes in a family with developmental delay and behavioural problems. BMC Med. Genet. 2012. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.J.; Rosenfeld, J.A.; Grant, E.; Gripp, K.W.; Anderson, C.; Aylsworth, A.S.; Saad, T.B.; Chizhikov, V.V.; Dybose, G.; Fagerberg, C.; et al. The duplication 17p13. 3 phenotype: Analysis of 21 families delineates developmental, behavioral and brain abnormalities, and rare variant phenotypes. Am. J. Med. Genet. 2013, 161, 1833–1852. [Google Scholar] [CrossRef] [PubMed]
- Grover, D.; Verma, R.; Goes, F.S.; Mahon, P.L.B.; Gershon, E.S.; McMahon, F.J.; Potash, J.B. Family based association of YWHAH in psychotic bipolar disorder. Am. J. Med. Genet. 2009, 150B, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z.Q.; Li, J.Y.; Li, T.; Wang, T.; Li, Y.; Xu, Y.F.; Feng, G.Y.; Shi, Y.Y.; He, L. Polymorphisms and haplotypes in the YWHAE gene increase susceptibility to bipolar disorder in Chinese Han population. J. Clin. Psychiatry 2012, 73, e1276–e1282. [Google Scholar] [CrossRef] [PubMed]
- Kido, M.; Nakamura, Y.; Nemoto, K.; Takahashi, T.; Aleksic, B.; Furuichi, A.; Nakamura, Y.; Ikeda, M.; Noguchi, K.; Kaibuchi, K.; et al. The Polymorphism of YWHAE, a Gene Encoding 14–3-3Epsilon, and Brain Morphology in Schizophrenia: A Voxel-Based Morphometric Study. PLoS ONE 2014, 9, e103571. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nakamura, Y.; Nakamura, Y.; Aleksic, B.; Takayanagi, Y.; Furuichi, A.; Kido, M.; Nakamura, M.; Sasabayashi, D.; Ikeda, M.; et al. The polymorphism of YWHAE, a gene encoding 14–3-3epsilon, and orbitofrontal sulcogyral pattern in patients with schizophrenia and healthy subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 51, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Bunney, T.D.; De Boer, A.H.; Levin, M. Fusicoccin signaling reveals 14–3-3 protein function as a novel step in left-right patterning during amphibian embryogenesis. Development 2003, 130, 4847–4858. [Google Scholar] [CrossRef] [PubMed]
- Nellist, M.; Goedbloed, M.A.; Halley, D.J.J. Regulation of tuberous sclerosis complex (TSC) function by 14–3-3 proteins. Biochem. Soc. Trans. 2003, 31, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Hengstschläger, M.; Rosner, M.; Fountoulakis, M.; Lubec, G. Tuberous sclerosis genes regulate cellular 14–3-3 protein levels. Biochem 2003, 312, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.S.; Ramshaw, H.S.; Thomas, P.Q.; Toyo-Oka, K.; Xu, X.; Martin, S.; Coyle, P.; Guthridge, M.A.; Stomski, F.; Van Den Buuse, M.; et al. Neurodevelopmental and neuropsychiatric behaviour defects arise from 14–3-3ζ deficiency. Mol. Psychiatry 2012, 17, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Taya, S.; Shinoda, T.; Tsuboi, D.; Asaki, J.; Nagai, K.; Hikita, T.; Kuroda, S.; Kuroda, K.; Shimizu, M.; Hirotsune, S.; et al. DISC1 regulates the transport of the NUDEL/LIS1/14–3-3ε complex through kinesin-1. J. Neurosci 2007, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Manbodh, R.; Sheppard, C.; Chalmers, A.D. RASSF7 is a member of a new family of RAS association domain–containing proteins and is required for completing mitosis. Mol. Biol. Cell 2008, 19, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoets, G.; Li, X.; Bogaert, S.; Roberts, N. Hand preference, cognitive performance, and brain asymmetry in situs inversus totalis. In Proceedings of North Sea Laterality Meeting 2016, Groningen, The Netherlands, 1–3 September 2016.
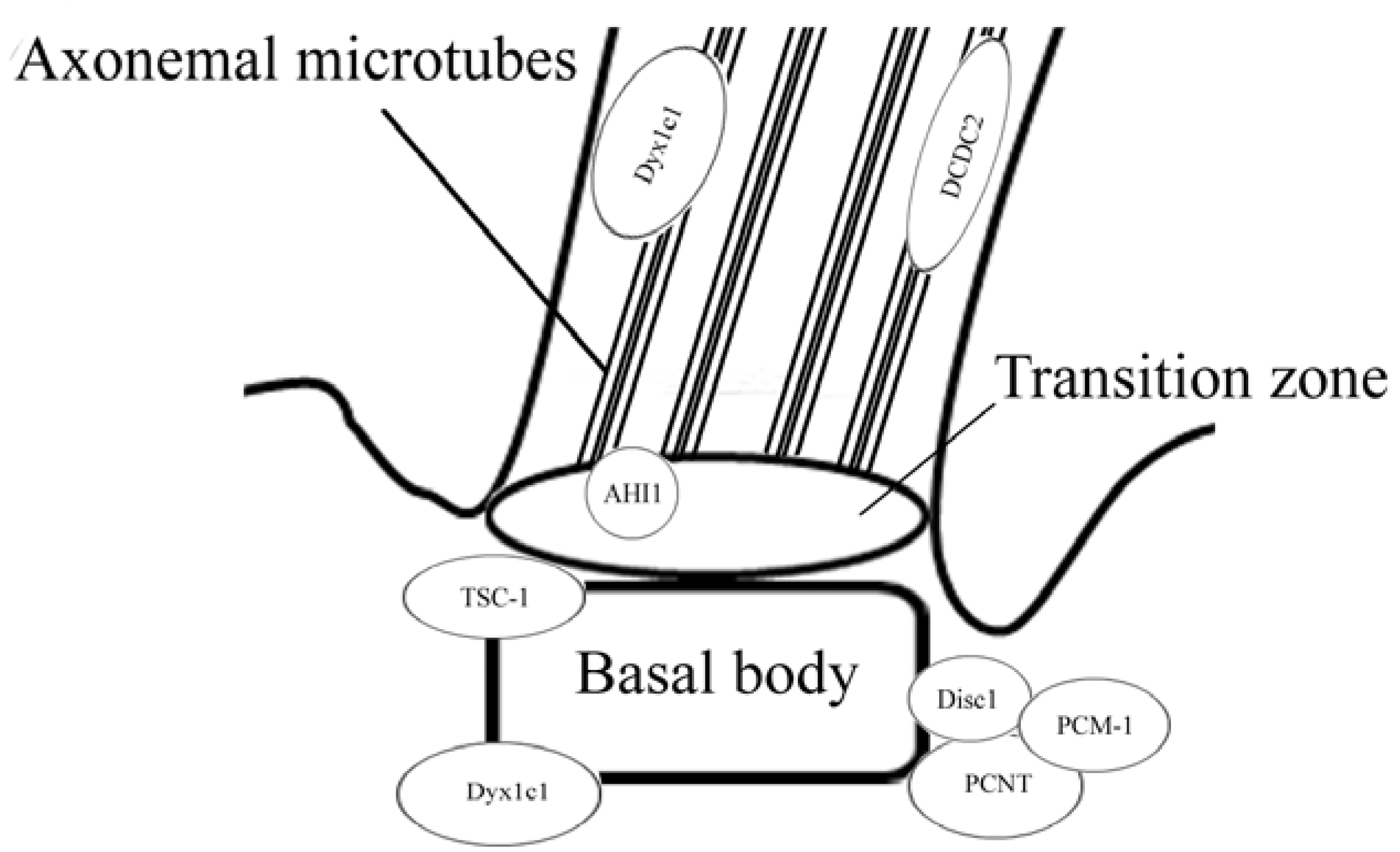
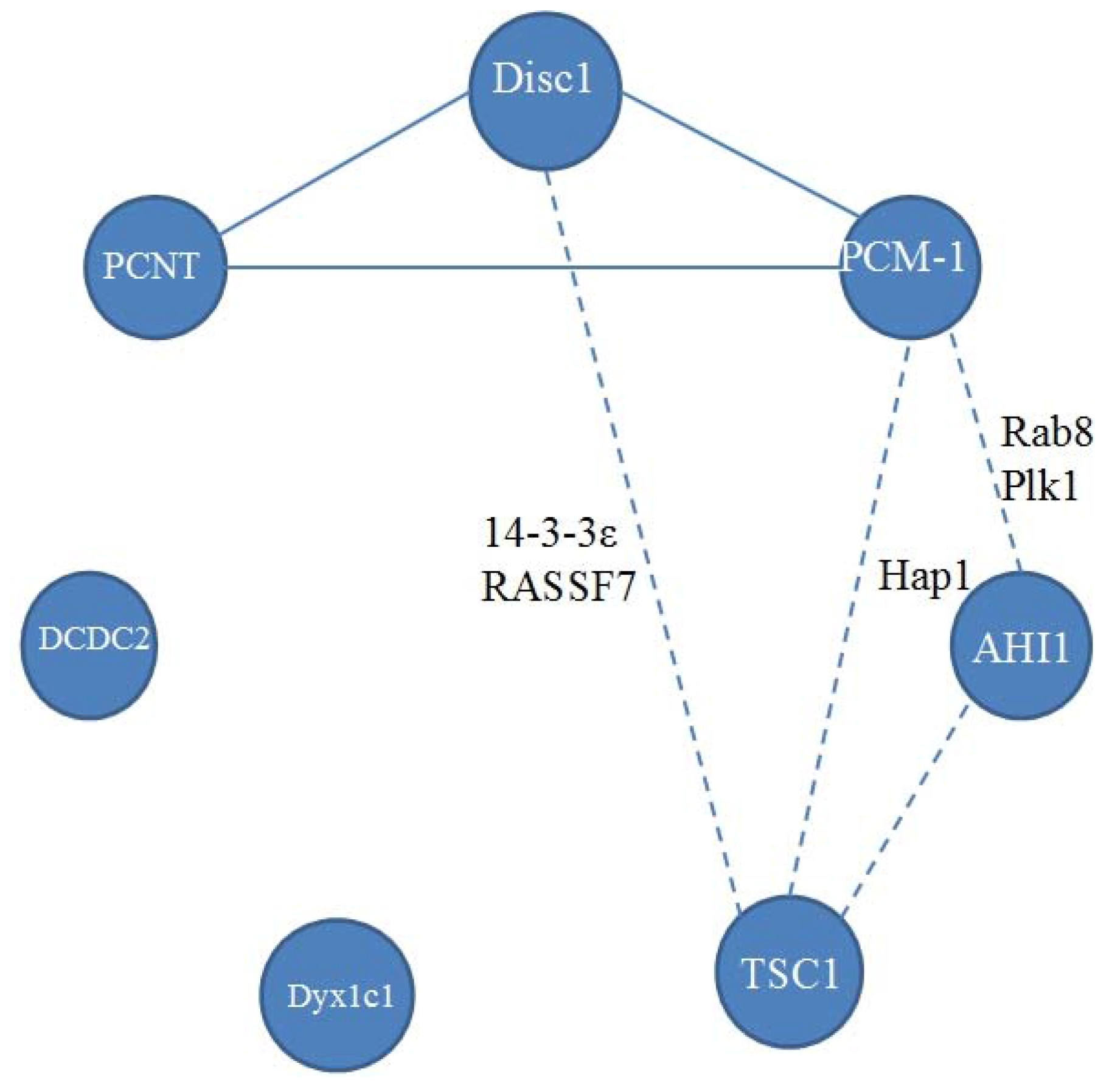
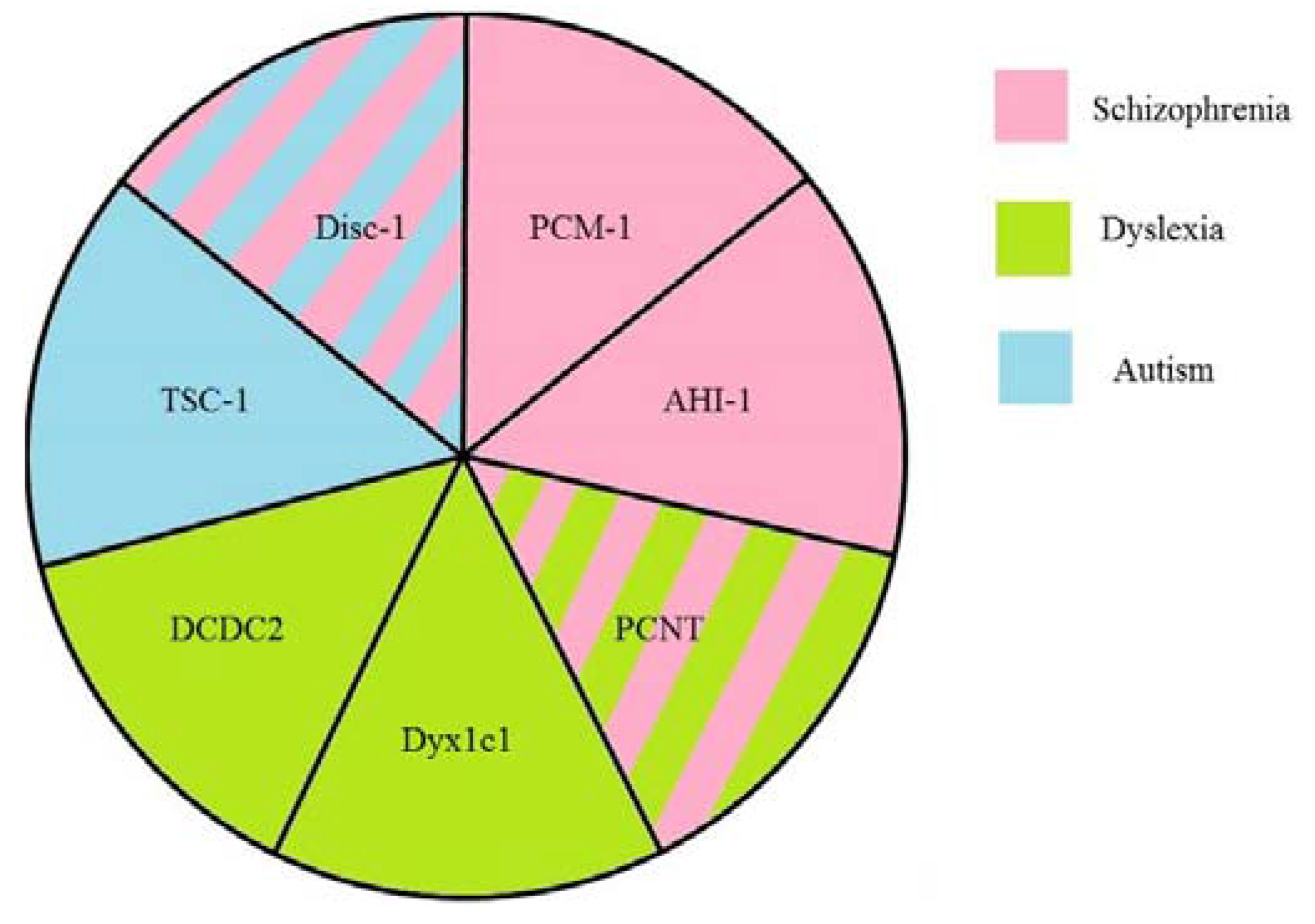

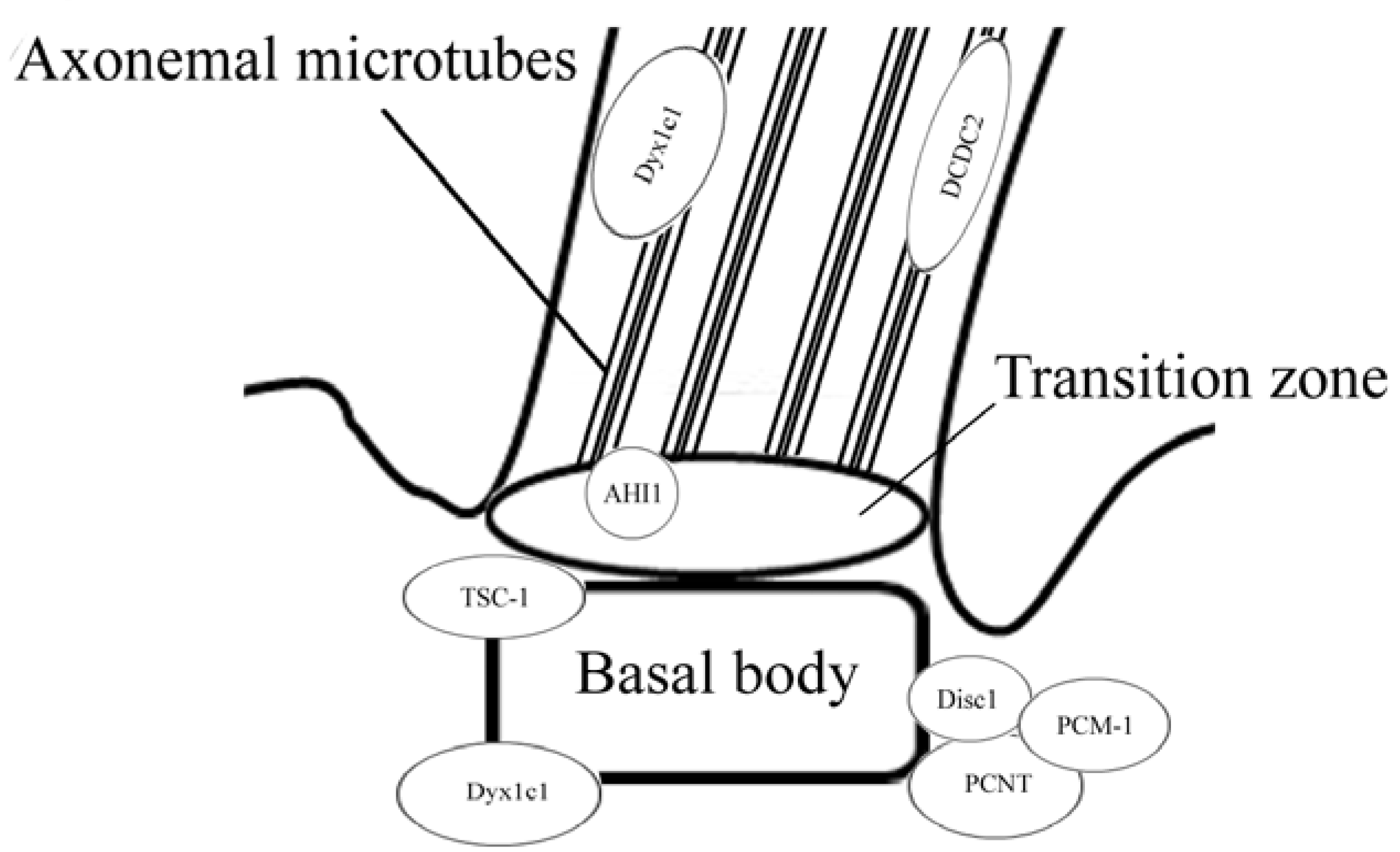{kind=link}
{kind=link}
{kind=link}
| Protein | Function in the Cilia | Other Functions | Involvement in Visceral Asymmetry | Associated Psychiatric Disorders | Suggested Mechanisms | Ref. |
|---|---|---|---|---|---|---|
| Disc1 | ciliogenesis and intraflagellar transport regulation | microtubular transport, probably mitochondrial protein import machinery, Akt/mTOR and GSK-3/β-catenin/Wnt pathways | schizophrenia, autism, depression, bipolar disorder | neuronal migration, neuronal signaling and signal transduction, axonal bundling, transport of GABA-containing vesicles | [49,50,51,52,53,54] | |
| PCM-1 | ciliogenesis and cilia disassembly | microtubule-based trafficking of proteins to the centrosome, centrosome assembly | heart left-right asymmetry in zebrafish | schizophrenia | cell cycle regulation and migration of neurons alone or in coordination with Disc1 | [55,56,57,58] |
| PCNT | interacts with proteins involved in cilia assembly | pericentriolar matrix assembly, anchors the γ-tubulin complex to the centrosome, providing microtubule nucleation sites | dyslexia schizophrenia | functioning of the centrosomes and the cytoskeleton, interneuron migration | [59,60,61] | |
| AHI1 | prevention of non-ciliarymembrane proteins from diffusing into the ciliary membrane, cilia assembly via interaction with Rab8a | traffic of endocytic vesicles | heart looping in zebrafish knockdown | schizophrenia bipolar disorder | in complex with Hap1 maintains the level of TrkB, neuronal migration | [62,63,64,65,66,67] |
| TSC1 | inhibits formation of the extra cilia | cell cycle regulation, methabolism, cell polarity, mTOR, PI3K-Akt, the ERK1/2-RSK1 signaling | affected expression of southpaw gene in zebrafish morphants | autism | maintenance of dendrite spine density, mTOR signaling pathway, neuronal migration | [68,69,70,71] |
| DCDC2 | ciliogenesis and ciliary signaling | promotes Shh signaling and inhibits Wnt signaling | left-right asymmetry defects in liver, gut, and pancreas | dyslexia | maintenance of the balance between Shh and Wnt signaling, neuronal migration | [13,42,72] |
| DYX1C1 | ciliogenesis and cilia motility (dynein arm assembly) | normal heart looping, left-right asymmetry defects in liver, gut, and pancreas | dyslexia | neuronal migration | [73,74,75,76] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trulioff, A.; Ermakov, A.; Malashichev, Y. Primary Cilia as a Possible Link between Left-Right Asymmetry and Neurodevelopmental Diseases. Genes 2017, 8, 48. https://doi.org/10.3390/genes8020048
Trulioff A, Ermakov A, Malashichev Y. Primary Cilia as a Possible Link between Left-Right Asymmetry and Neurodevelopmental Diseases. Genes. 2017; 8(2):48. https://doi.org/10.3390/genes8020048
Chicago/Turabian StyleTrulioff, Andrey, Alexander Ermakov, and Yegor Malashichev. 2017. "Primary Cilia as a Possible Link between Left-Right Asymmetry and Neurodevelopmental Diseases" Genes 8, no. 2: 48. https://doi.org/10.3390/genes8020048
APA StyleTrulioff, A., Ermakov, A., & Malashichev, Y. (2017). Primary Cilia as a Possible Link between Left-Right Asymmetry and Neurodevelopmental Diseases. Genes, 8(2), 48. https://doi.org/10.3390/genes8020048