
Development of Gene?Based SSR Markers in Winged Bean (Psophocarpus tetragonolobus (L.) DC.) for Diversity Assessment

Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material, RNA Extraction, Complementary DNA (cDNA) Library Construction, and Sequencing
2.2. De Novo Transcriptome Assembly and Microsatellite Identification
2.3. Microsatellite Markers Development and Scoring
2.4. Cluster Analysis
3. Results and Discussion
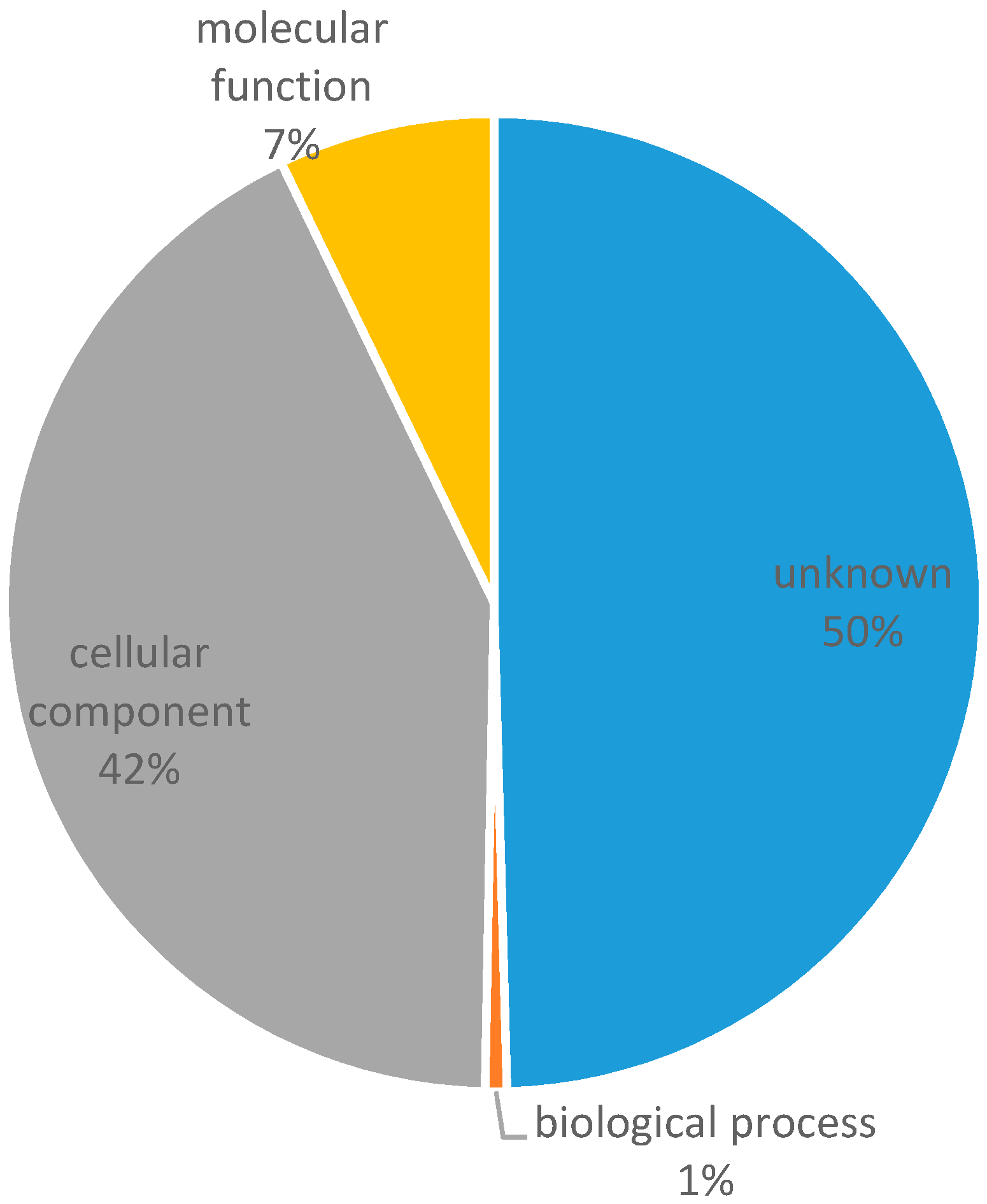
3.1. Transcriptome Assembly and In Silico Identification of Microsatellites
3.2 Development of SSR Markers and Cluster Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- NAS. The Winged Bean: High-Protein Crop for the Humid Tropics, 2nd ed.; National Academy Press: Washington, DC, USA, 1981. [Google Scholar]
- Klu, G.Y.P. Induced mutations for accelerated domestication-a case study of winged bean. West Afr. J. Appl. Ecol. 2000, 1, 47–52. [Google Scholar]
- Khan, T.N. Papua New Guinea: A centre of genetic diversity in winged bean (Psophocarpus tetragonologus (L.) Dc.). Euphytica 1976, 25, 693–705. [Google Scholar] [CrossRef]
- Harder, D.K. Chromosome Counts in Psophocarpus. Kew Bull. 1992, 47, 529–534. [Google Scholar] [CrossRef]
- Harder, D.K.; Smartt, J. Further evidence on the origin of the cultivated winged bean, Psophocarpus tetragonolobus (L.) DC. Econ. Bot. 1992, 46, 187–191. [Google Scholar] [CrossRef]
- Prakash, D.; Misra, P.N.; Misra, P.S. Amino acid profile of winged bean (Psophocarpus tetragonolobus (L.) DC.): A rich source of vegetable protein. Plant Foods Hum. Nutr. 1987, 37, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.S.; Salunkhe, D.K. Winged bean in human nutrition. Crit. Rev. Food Sci. Nutr. 1984, 21, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Okezie, B.O.; Martin, F.W. Chemical composition of dry seeds and fresh leaves of winged bean varieties grown in the U.S. and Puerto Rico. J. Food Sci. 1980, 45, 1045–1051. [Google Scholar] [CrossRef]
- Anugroho, F.; Kitou, M.; Kinjo, K.; Kobashigawa, N. Growth and nutrient accumulation of winged bean and velvet bean as cover crops in a subtropical region. Plant Prod. Sci. 2010, 13, 360–366. [Google Scholar] [CrossRef]
- Banerjee, A.; Bagchi, D.K.; Si, L.K. Studies on the potential of winged bean as a multipurpose legume cover crop in tropical regions. Exp. Agric. 2008, 20, 297–301. [Google Scholar] [CrossRef]
- Hikam, S.; MacKown, C.T.; Poneleit, C.G.; Hildebrand, D.F. Growth and N accumulation in maize and winged bean as affected by N level and intercropping. Ann. Bot. 1991, 68, 17–22. [Google Scholar] [CrossRef]
- FAO. Coping with Climate Change—The Roles of Genetic Resources for Food and Agriculture; FAO: Rome, Italy, 2015. [Google Scholar]
- Mohanty, C.S.; Verma, S.; Singh, V.; Khan, S.; Gaur, P.; Gupta, P.; Nizar, M.A.; Dikshit, N.; Pattanayak, R.; Shukla, A.; et al. Characterization of winged bean (Psophocarpus tetragonolobus (L.) DC.) based on molecular, chemical and physiological parameters. Am. J. Mol. Biol. 2013, 3, 187–197. [Google Scholar] [CrossRef]
- Koshy, E.P.P.; Alex, B.K.K.; John, P. Clonal fidelity studies on regenerants of Psophocarpus tetragonolobus (L.) DC. using RAPD markers. Bioscan 2013, 8, 763–766. [Google Scholar]
- Chen, D.; Yi, X.; Yang, H.; Zhou, H.; Yu, Y.; Tian, Y.; Lu, X. Genetic diversity evaluation of winged bean (Psophocarpus tetragonolobus (L.) DC.) using inter-simple sequence repeat (ISSR). Genet. Resour. Crop Evol. 2015, 62, 823–828. [Google Scholar] [CrossRef]
- Vatanparast, M.; Shetty, P.; Chopra, R.; Doyle, J.J.; Sathyanarayana, N.; Egan, A.N. Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Sci. Rep. 2016, 6, 29070. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A. Transcriptome sequencing and marker development for four underutilized legumes. Appl. Plant Sci. 2015, 3, 1400111. [Google Scholar] [CrossRef] [PubMed]
- Erskine, W. Measurements of the cross-pollination of winged bean in Papua New Guinea. SABRAO J. Breed. Genet. 1980, 12, 11–14. [Google Scholar]
- Eagleton, G.E. Evaluation of Genetic Resources in the Winged Bean (Psophocarpus tetragonolobus (L.) DC.) and Their Utilisation in the Development of Cultivars for Higher Latitudes. Ph.D. Dissertation, University of Western Australia, Perth, Australia, 1983. [Google Scholar]
- De Silva, H.N.; Omran, A. Diallel analysis of yield and yield components of winged bean (Psophocarpus tetragonolobus (L.) D.C.). J. Agric. Sci. 1986, 106, 485–490. [Google Scholar] [CrossRef]
- Erskine, W.; Khan, T.N. Inheritance of pigmentation and pod shape in winged bean. Euphytica 1977, 26, 829–831. [Google Scholar] [CrossRef]
- Erskine, B.Y.W. Heritability and combining ability of vegetative and phenological characters of winged beans (Psophocarpus tetragonolobus (L.) DC.). J. Agric. Sci. 1981, 96, 503–508. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, version 1.33; Software; 2011. Available online: https://github.com/najoshi/sickle (accessed on 13 March 2013).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32 (Suppl. 1), D258–D261. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. DNA Protocols for Plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- VSN International. GenStat for Windows, 18th ed.; VSN International: Hemel Hempstead, UK, 2015. [Google Scholar]
- Jayashree, B.; Punna, R.; Prasad, P.; Bantte, K.; Hash, C.T.; Chandra, S.; Hoisington, D.A.; Varshney, R.K. A database of simple sequence repeats from cereal and legume expressed sequence tags mined in silico: Survey and evaluation. In Silico Biol. 2006, 6, 607–620. [Google Scholar] [PubMed]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K.; et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Bansal, R.; Gopalakrishna, T. Development and characterization of genic SSR markers for mungbean (Vigna radiata (L.) Wilczek). Euphytica 2014, 195, 245–258. [Google Scholar] [CrossRef]
- Blair, M.W.; Hurtado, N.; Chavarro, C.M.; Muñoz-Torres, M.C.; Giraldo, M.C.; Pedraza, F.; Tomkins, J.; Wing, R.; Varshney, R.; Graner, A.; et al. Gene-based SSR markers for common bean (Phaseolus vulgaris L.) derived from root and leaf tissue ESTs: An integration of the BMc series. BMC Plant Biol. 2011, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Gopalakrishna, T. Development of unigene-derived SSR markers in cowpea (Vigna unguiculata) and their transferability to other Vigna species. Genome 2010, 53, 508–523. [Google Scholar] [PubMed]
- Molosiwa, O.O.; Aliyu, S.; Stadler, F.; Mayes, K.; Massawe, F.; Kilian, A.; Mayes, S. SSR marker development, genetic diversity and population structure analysis of Bambara groundnut [Vigna subterranean (L.) Verdc.] landraces. Genet. Resour. Crop Evol. 2015, 62, 1225–1243. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Individuals | Origin |
|---|---|
| Tpt53-9-8 Tpt53-9-10 | Bangladesh |
| Tpt17-6-3 Tpt17-6-8 | Indonesia |
| M3-3 | Malaysia |
| Tpt10-7-5 Tpt10-7-7 | Papua New Guinea |
| SLS319-10-3 SLS319-10-4 | Sri Lanka |
| Tissue | Leaf | Pod | Reproductive Tissue | Root |
|---|---|---|---|---|
| Number of raw read/base (bp) | 3,150,356/1,544,004,822 | 3,973,092/1,868,456,680 | 3,544,968/1,719,303,632 | 3,873,893/1,859,527,511 |
| Numbers of trimmed read/base (bp) | 3,113,502/1,438,258,180 | 3,157,832/1,301,766,113 | 3,199,527/1,431,024,141 | 3,303,324/1,461,908,151 |
| Number of contigs/base (bp) | 198,554/158,382,439 | |||
| Average contig size (bp) | 798 | |||
| N50 | 1462 | |||
| Total number of sequences examined | 198,554 |
| Total size of examined sequences (bp) | 158,382,439 |
| Total number of identified SSRs | 9682 |
| Number of SSR containing sequences | 8793 |
| Number of sequences containing more than one SSR | 780 |
| Number of SSRs present in compound formation | 352 |
| Number of dimer-repeat | 4500 |
| Number of trimer-repeat | 4855 |
| Number of tetramer-repeat | 279 |
| Number of pentamer-repeat | 48 |
| Number of Repeat Motif | Total | % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Di-nucleotide | 5 | 6 | 7 | 8 | 9 | 10 | >10 | ||
| AC/GT/CA/TG | - | 256 | 138 | 63 | 37 | 12 | 10 | 516 | 11.5 |
| AG/CT/GA/TC | - | 995 | 543 | 330 | 391 | 434 | 189 | 2882 | 64.0 |
| AT/TA | - | 407 | 201 | 1167 | 113 | 107 | 68 | 1063 | 23.6 |
| CG/GC | - | 38 | 1 | 0 | 0 | 0 | 0 | 39 | 0.9 |
| Total | - | 1696 (37.7%) | 883 (19.6%) | 560 (12.4%) | 541 (12.0%) | 553 (12.3%) | 267 (5.9%) | 4500 | |
| Tri-nucleotide | |||||||||
| AAC/ACA/CAA/GTT/TGT/TTG | 279 | 131 | 50 | 17 | 0 | 0 | 0 | 477 | 9.8 |
| AAG/AGA/GAA/CTT/TCT/TTC | 612 | 405 | 351 | 11 | 0 | 0 | 0 | 1379 | 28.4 |
| AAT/ATA/TAA/TTA/TAT/ATT | 305 | 145 | 112 | 15 | 0 | 0 | 0 | 577 | 11.9 |
| ACC/CAC/CCA/GGT/GTG/TGG | 307 | 62 | 55 | 10 | 0 | 0 | 0 | 434 | 8.9 |
| ACG/CGA/GAC/CGT/GTC/TCG | 90 | 66 | 12 | 7 | 0 | 0 | 0 | 175 | 3.6 |
| ACT/CTA/TAC/AGT/TAG/GTA | 36 | 8 | 3 | 3 | 0 | 0 | 0 | 50 | 1.0 |
| AGC/CAG/GCA/TGC/CTG/GCT | 271 | 105 | 38 | 9 | 0 | 0 | 0 | 423 | 8.7 |
| AGG/GGA/GAG/TCC/CTC/CCT | 247 | 115 | 75 | 11 | 0 | 0 | 0 | 448 | 9.2 |
| ATC/CAT/TCA/GAT/ATG/TGA | 311 | 83 | 24 | 34 | 0 | 0 | 0 | 452 | 9.3 |
| CCG/CGC/GCC/GGC/GCG/CGG | 247 | 130 | 55 | 8 | 0 | 0 | 0 | 440 | 9.1 |
| Total | 2705 (55.7%) | 1250 (25.7%) | 775 (16.0%) | 125 (2.6%) | 0 | 0 | 0 | 4855 | |
| Marker | Papua New Guinea | Indonesia | Bangladesh | Sri Lanka | Malaysia | ||||
|---|---|---|---|---|---|---|---|---|---|
| Tpt10-7-5 | Tpt10-7-7 | Tpt17-6-3 | Tpt17-6-8 | Tpt53-9-8 | Tpt53-9-10 | SLS319-10-3 | SLS319-10-4 | M3-3 | |
| P27.2 | 205 | 199/205 | 199 | 205 | 205 | 205 | 205 | 205 | 205 |
| P43.2 | 199 | 199 | 195 | 195 | 197 | 199 | 199 | 199 | 195 |
| Pt1.1 | 335 | 335 | 339 | 339 | 339 | 335/339 | 335 | 335 | 339 |
| Pt10 | 226/228 | 228 | 226 | 226 | 228 | 228 | 228 | 228 | 228 |
| Pt14 | 358 | 358 | 352 | 352 | 350 | 350 | 358 | 358 | 354 |
| Pt24 | 219 | 217/219 | 217 | 217 | 219 | 219 | 219 | 219 | 217 |
| Pt7.2 | 426/432 | 426 | 426 | 426 | 428 | 428 | 426 | 426 | 426/428 |
| WB17 | 198 | 198 | 198 | 198 | 198 | 194/198 | 198 | 198 | 198 |
| Pt53 | 315 | 309 | 315 | 315 | 309/315 | 309/315 | 312 | 312 | 315 |
| Pt58 | 255/261 | 255/261 | 261 | 261 | 261 | 261 | 261 | 261 | 261 |
| Pt65.1 | 273 | 273 | 267 | 267 | 267 | 267 | 267 | 267 | 267/273 |
| Pt67.1 | 293 | 293 | 296 | 296 | 293 | 293 | 296 | 296 | 293/296 |
| Pt68.1 | 226 | 226 | 229 | 229 | 226 | 223/226 | 223 | 223/226 | 226/235 |
| Pt76.1 | 203 | 203 | 203 | 203 | 209 | 209 | 209 | 209 | 209 |
| Pt78.1 | 306/309 | 306/309 | 306 | 306 | 309 | 309 | 306 | 306 | 309 |
| Pt85.1 | 276/279 | 276/279 | 276 | 276 | 276 | 276 | 276 | 276 | 279 |
| Pt93.1 | 266 | 266 | 272 | 272 | 266/272 | 272 | 266 | 266 | 276 |
| Pt99.2 | 189/195 | 189/195 | 195 | 195 | 189 | 189 | 189 | 189 | 195 |
| Marker | SSR Motif | Major Allele Frequency | No. of Alleles | Heterozygosity | PIC |
|---|---|---|---|---|---|
| P27.2 | TA | 0.83 | 2 | 0.11 | 0.24 |
| P43.2 | TA | 0.56 | 3 | 0 | 0.49 |
| Pt1.1 | CT | 0.5 | 2 | 0.11 | 0.38 |
| Pt10 | TC | 0.72 | 2 | 0.11 | 0.32 |
| Pt14 | TG | 0.44 | 4 | 0 | 0.64 |
| Pt24 | GT | 0.61 | 2 | 0.11 | 0.36 |
| Pt7.2 | TC | 0.67 | 3 | 0.22 | 0.4 |
| WB17 | GA | 0.94 | 2 | 0.11 | 0.1 |
| Average dimer SSR markers | 0.66 | 2.5 | 0.1 | 0.37 | |
| Pt53 | CGC | 0.56 | 3 | 0.22 | 0.53 |
| Pt58 | TAG | 0.89 | 2 | 0.22 | 0.18 |
| Pt65.1 | CAG | 0.72 | 2 | 0.11 | 0.32 |
| Pt67.1 | AGA | 0.5 | 2 | 0.11 | 0.38 |
| Pt68.1 | AAC | 0.5 | 4 | 0.33 | 0.59 |
| Pt76.1 | CGC | 0.56 | 2 | 0 | 0.37 |
| Pt78.1 | AAC | 0.56 | 2 | 0.22 | 0.37 |
| Pt85.1 | GCG | 0.78 | 2 | 0.22 | 0.29 |
| Pt93.1 | TGT | 0.5 | 3 | 0.11 | 0.5 |
| Pt99.2 | TTC | 0.56 | 2 | 0.22 | 0.37 |
| Average trimer SSR marker | 0.61 | 2.4 | 0.18 | 0.39 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, Q.N.; Tanzi, A.S.; Ho, W.K.; Malla, S.; Blythe, M.; Karunaratne, A.; Massawe, F.; Mayes, S. Development of Gene?Based SSR Markers in Winged Bean (Psophocarpus tetragonolobus (L.) DC.) for Diversity Assessment. Genes 2017, 8, 100. https://doi.org/10.3390/genes8030100
Wong QN, Tanzi AS, Ho WK, Malla S, Blythe M, Karunaratne A, Massawe F, Mayes S. Development of Gene?Based SSR Markers in Winged Bean (Psophocarpus tetragonolobus (L.) DC.) for Diversity Assessment. Genes. 2017; 8(3):100. https://doi.org/10.3390/genes8030100
Chicago/Turabian StyleWong, Quin Nee, Alberto Stefano Tanzi, Wai Kuan Ho, Sunir Malla, Martin Blythe, Asha Karunaratne, Festo Massawe, and Sean Mayes. 2017. "Development of Gene?Based SSR Markers in Winged Bean (Psophocarpus tetragonolobus (L.) DC.) for Diversity Assessment" Genes 8, no. 3: 100. https://doi.org/10.3390/genes8030100
APA StyleWong, Q. N., Tanzi, A. S., Ho, W. K., Malla, S., Blythe, M., Karunaratne, A., Massawe, F., & Mayes, S. (2017). Development of Gene?Based SSR Markers in Winged Bean (Psophocarpus tetragonolobus (L.) DC.) for Diversity Assessment. Genes, 8(3), 100. https://doi.org/10.3390/genes8030100