
Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Clinical Features of FSHD
1.2. The DUX4 Gene and Protein
1.3. The Genetic and Epigenetic Conditions Required to Develop FSHD
1.4. Therapeutic Approaches
2. Material and Methods
2.1. Ethics Statement
2.2. Myogenic Cell Culture
2.3. siRNA and AOs Transfection
2.4. Immunofluorescence
2.5. RNA Analysis
2.6. Statistical Analyses
3. Results and Discussion
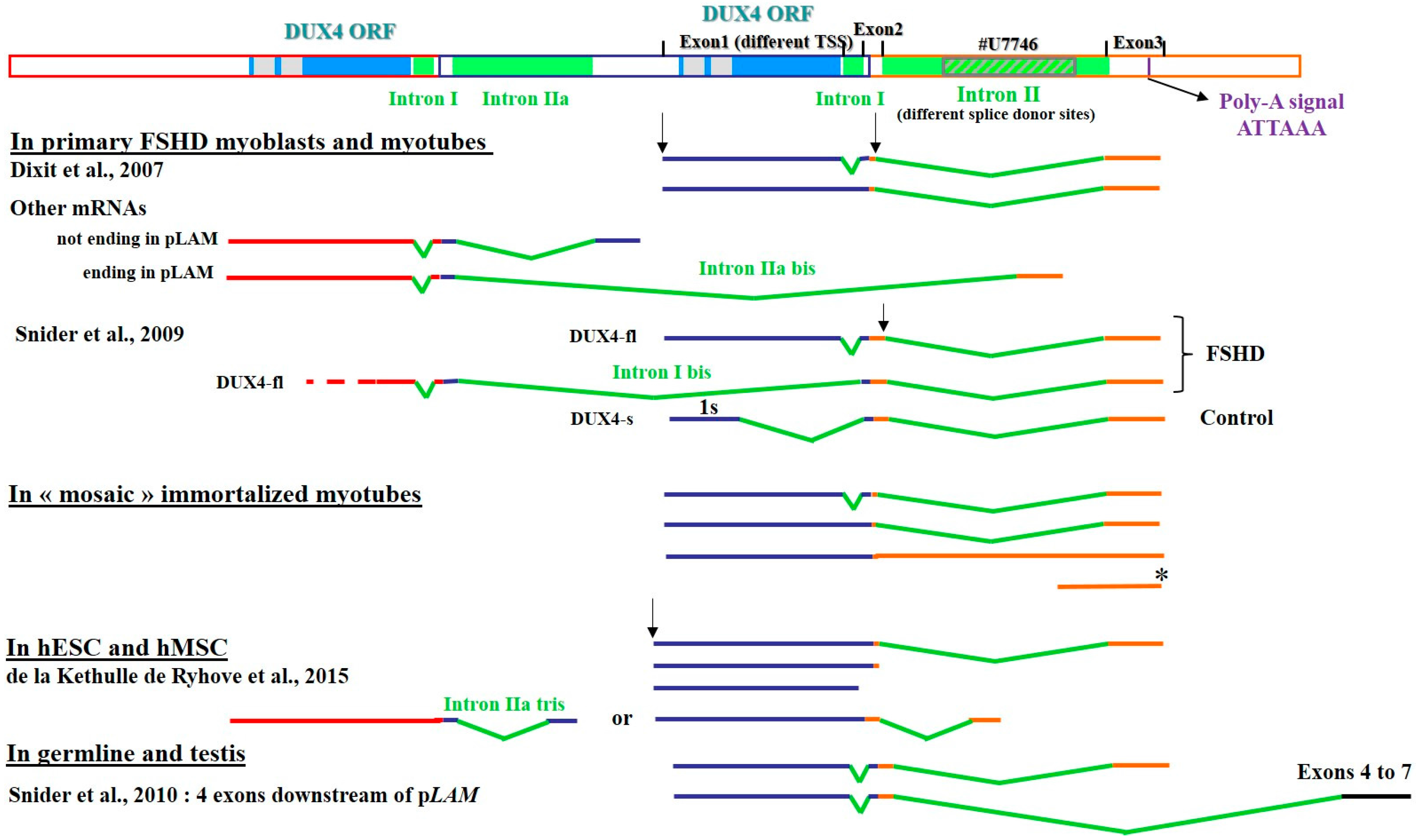
3.1. The Heterogeneity of DUX4 Transcripts
3.2. Defining Targets for Antisense Oligonucleotides on DUX4 mRNAs
3.2.1. Interference with mRNA Cleavage and Polyadenylation
3.2.2. Interference with mRNA Splicing
Muscle Cells
Human Mesenchymal Stromal Cells and Embryonic Stem Cells
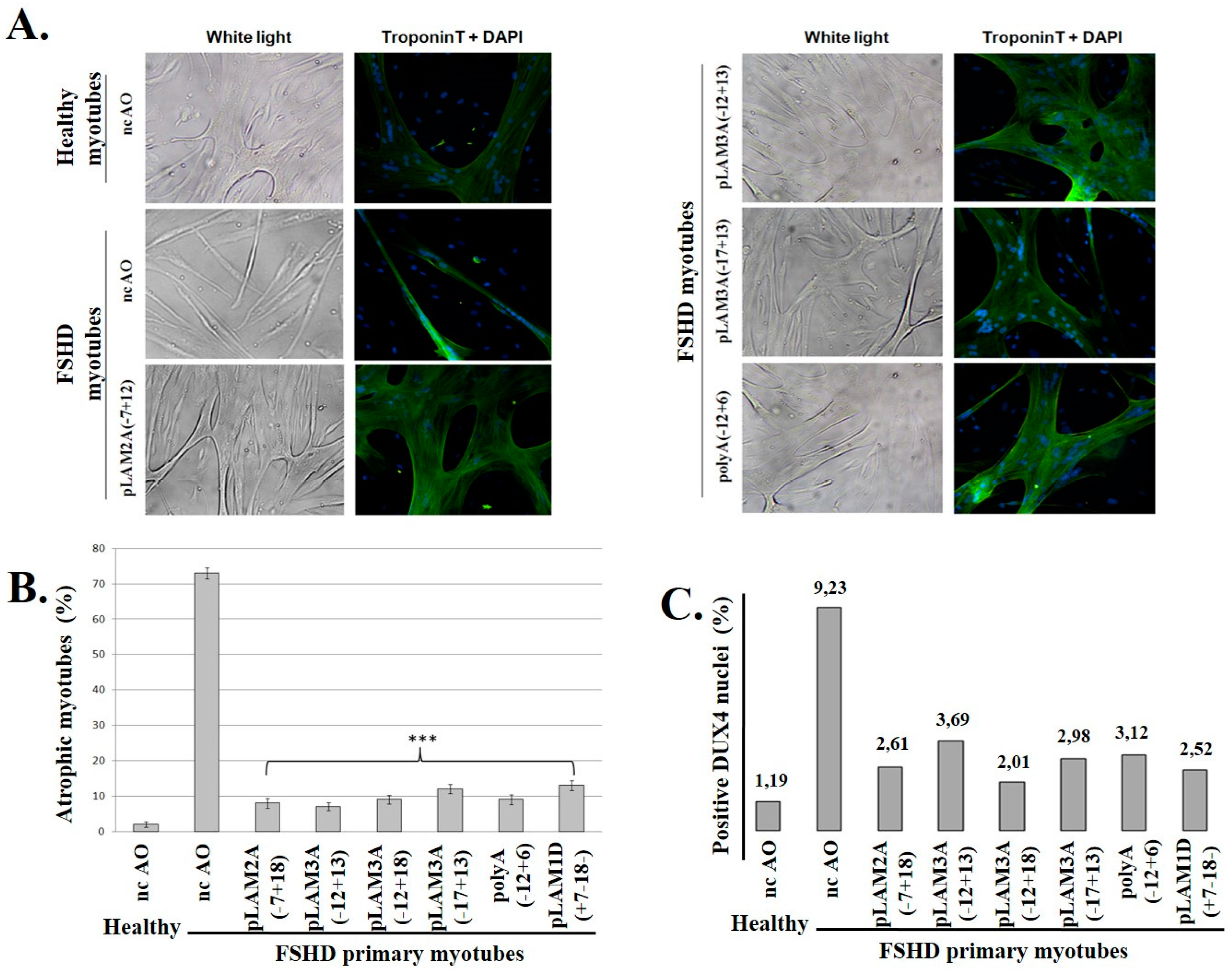
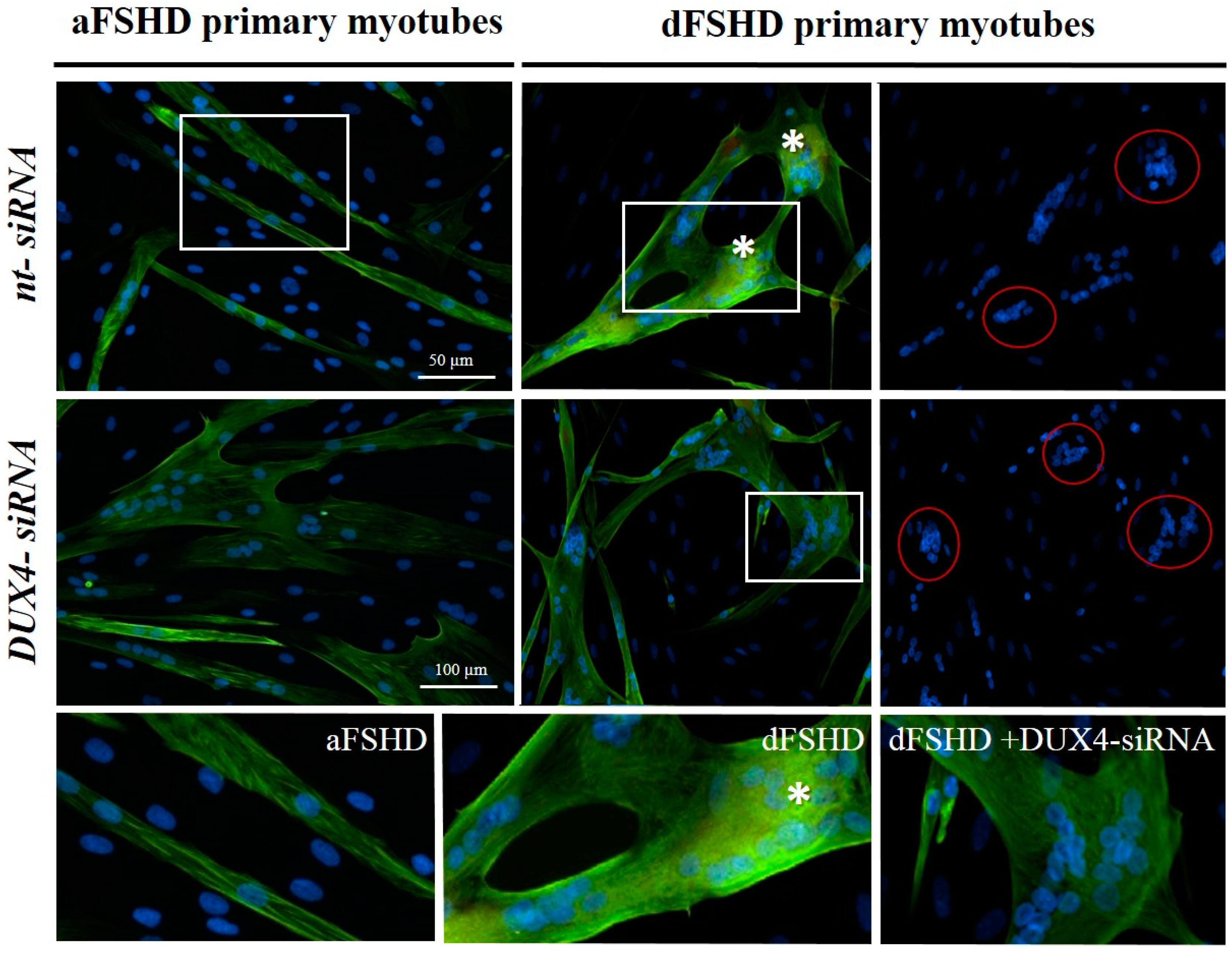
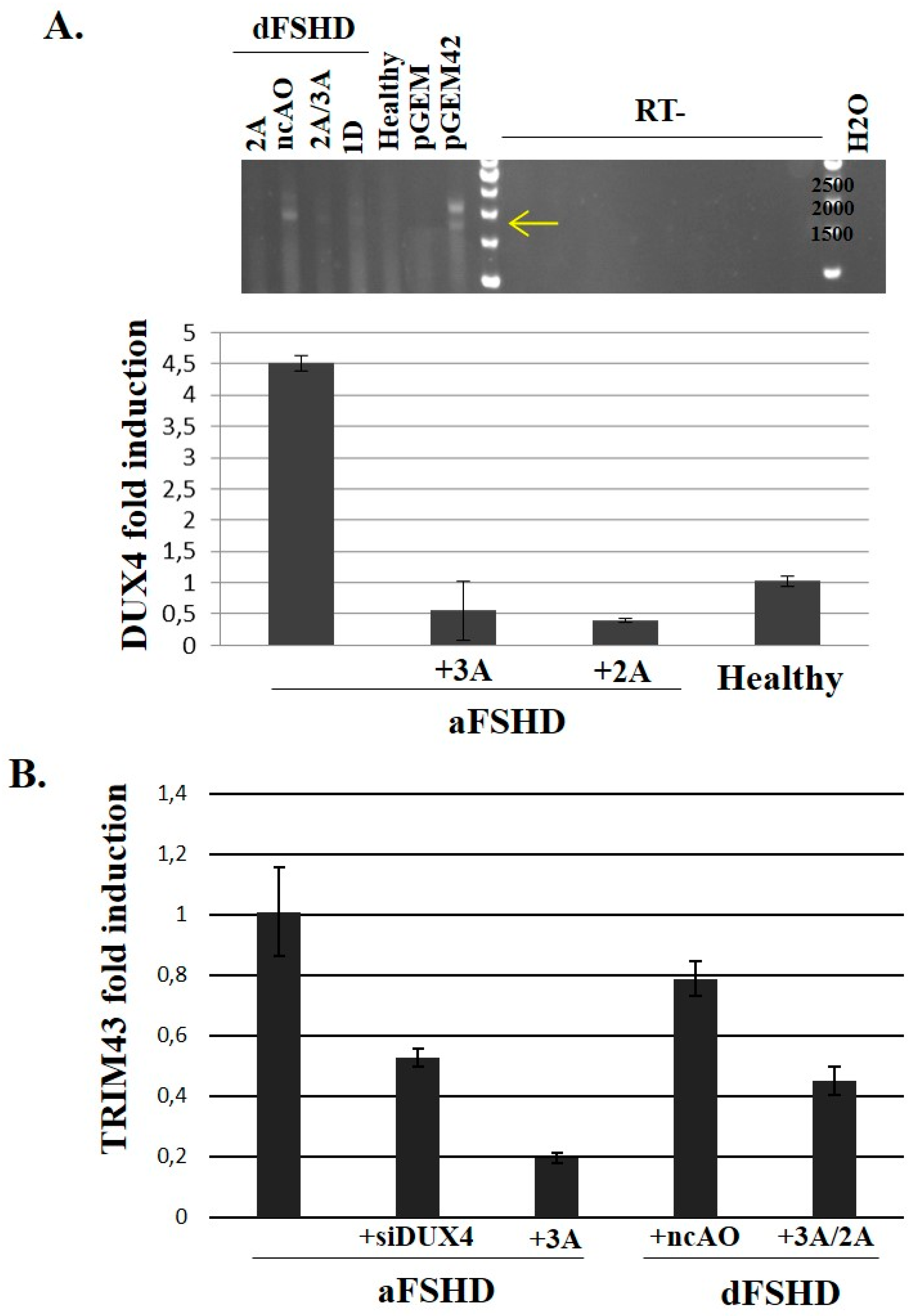
3.3. DUX4 Inhibition Prevents the Formation of Atrophic, but Not Disorganized FSHD Myotubes
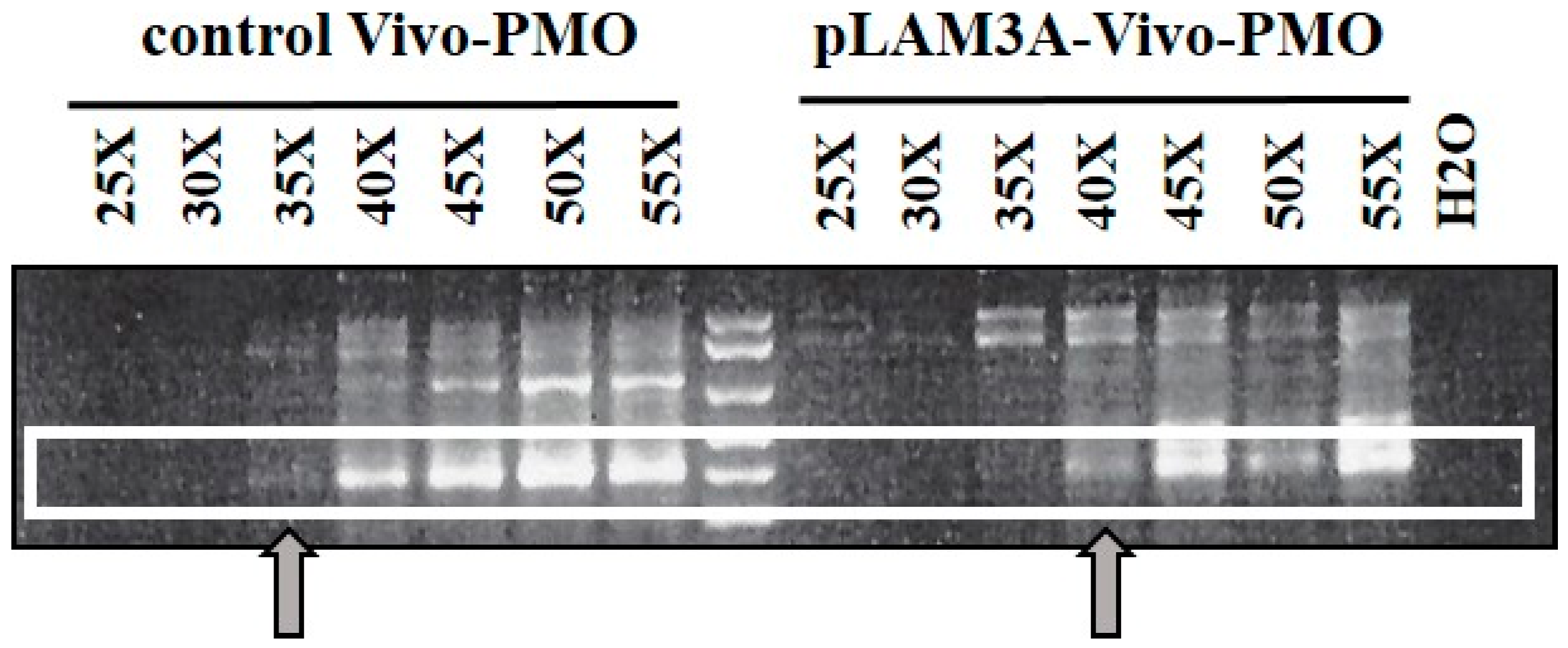
3.4. Evaluation of AOs in Preclinical Models
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Orpha.net. The portal for rare diseases and orphan drugs. Available online: http://www.orpha.net (accessed on 24 February 2017).
- Deenen, J.C.W.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; van Engelen, B.G.M. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Mul, K.; Lassche, S.; Voermans, N.C.; Padberg, G.W.; Horlings, C.G.; van Engelen, B.G. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pract. Neurol. 2016, 16, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; Raynaud de Mauverger, E.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Neurol. Clin. 2014, 32, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Kilmer, D.D.; Abresch, R.T.; McCrory, M.A.; Carter, G.T.; Fowler, W.M.; Johnson, E.R.; McDonald, C.M. Profiles of neuromuscular diseases. Facioscapulohumeral muscular dystrophy. Am. J. Phys. Med. Rehabil. 1995, 74, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Tasca, G.; Pescatori, M.; Monforte, M.; Mirabella, M.; Iannaccone, E.; Frusciante, R.; Cubeddu, T.; Laschena, F.; Ottaviani, P.; Ricci, E. Different molecular signatures in magnetic resonance imaging-staged facioscapulohumeral muscular dystrophy muscles. PLoS ONE 2012, 7, e38779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasca, G.; Monforte, M.; Ottaviani, P.; Pelliccioni, M.; Frusciante, R.; Laschena, F.; Ricci, E. Magnetic resonance imaging in a large cohort of facioscapulohumeral muscular dystrophy patients: Pattern refinement and implications for clinical trials. Ann. Neurol. 2016, 79, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.H.; Voet, N.B.M.; Nabuurs, C.I.; Kan, H.E.; de Rooy, J.W.J.; Geurts, A.C.; Padberg, G.W.; van Engelen, B.G.M.; Heerschap, A. Distinct disease phases in muscles of facioscapulohumeral dystrophy patients identified by MR detected fat infiltration. PLoS ONE 2014, 9, e85416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Della Marca, G.; Frusciante, R.; Scatena, M.; Dittoni, S.; Testani, E.; Vollono, C.; Losurdo, A.; Scarano, E.; Colicchio, S.; Farina, B.; et al. Heart rate variability in facioscapulohumeral muscular dystrophy. Funct. Neurol. 2010, 25, 211–216. [Google Scholar] [PubMed]
- Van Dijk, G.P.; van der Kooi, E.; Behin, A.; Smeets, J.; Timmermans, J.; van der Maarel, S.; Padberg, G.; Voermans, N.; van Engelen, B. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct. Neurol. 2014, 29, 159–165. [Google Scholar] [PubMed]
- Beckers, M.; Gabriëls, J.; van der Maarel, S.; De Vriese, A.; Frants, R.R.; Collen, D.; Belayew, A. Active genes in junk DNA? Characterization of DUX genes embedded within 3.3 kb repeated elements. Gene 2001, 264, 51–57. [Google Scholar] [CrossRef]
- Wijmenga, C.; Frants, R.R.; Brouwer, O.F.; Moerer, P.; Weber, J.L.; Padberg, G.W. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990, 336, 651–653. [Google Scholar] [CrossRef]
- Winokur, S.T.; Bengtsson, U.; Feddersen, J.; Mathews, K.D.; Weiffenbach, B.; Bailey, H.; Markovich, R.P.; Murray, J.C.; Wasmuth, J.J.; Altherr, M.R. The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: Implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res. 1994, 2, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.E.; Lyle, R.; Clark, L.N.; Valleley, E.M.; Wright, T.J.; Wijmenga, C.; van Deutekom, J.C.; Francis, F.; Sharpe, P.T.; Hofker, M. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 1994, 3, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Beckers, M.C.; Plaisance, S.; Marynen, P.; Collen, D.; Belayew, A. Characterization of a double homeodomain protein (DUX1) encoded by a cDNA homologous to 3.3 kb dispersed repeated elements. Hum. Mol. Genet. 1998, 7, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Gabriëls, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements and immune-mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Mattéotti, C.; Arias, C.; Corona, E.D.; Nuñez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Asawachaicharn, A.; Tyler, A.E.; Geng, L.N.; Petek, L.M.; Maves, L.; Miller, D.G.; Lemmers, R.J.L.F.; Winokur, S.T.; Tawil, R.; et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: New candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 2009, 18, 2414–2430. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, J.K.; Toso, E.A.; Lee, J.S.; Choi, S.H.; Slattery, M.; Aihara, H.; Kyba, M. DNA-binding sequence specificity of DUX4. Skelet. Muscle 2016, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Lamb, S.; Simsek, T.; Xu, Z.; Belayew, A.; Perlingeiro, R.; Kyba, M. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 2008, 214, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppée, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS ONE 2011, 6, e26820. [Google Scholar] [CrossRef] [PubMed]
- Knopp, P.; Krom, Y.D.; Banerji, C.R.S.; Panamarova, M.; Moyle, L.A.; den Hamer, B.; van der Maarel, S.M.; Zammit, P.S. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 2016, 129, 3816–3831. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Snider, L.; Balog, J.; Lemmers, R.J.L.F.; Van Der Maarel, S.M.; Tawil, R.; Tapscott, S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014, 23, 5342–5352. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Whiddon, J.L.; Yao, Z.; Kasinathan, B.; Snider, L.; Geng, L.N.; Balog, J.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013, 9, e1003947. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.; Shadle, S.C.; Resnick, R.; Snider, L.; Tawil, R.N.; van der Maarel, S.M.; Bradley, R.K.; Tapscott, S.J. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum. Mol. Genet. 2016, 25, 4419–4431. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Snider, L.; Jagannathan, S.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J.; Bradley, R.K. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. eLife 2015, 4, e04996. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Tsumagari, K.; Chang, S.-C.; Lacey, M.; Baribault, C.; Chittur, S.V.; Sowden, J.; Tawil, R.; Crawford, G.E.; Ehrlich, M. Gene expression during normal and FSHD myogenesis. BMC Med. Genom. 2011, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Beermann, M.L.; Boyce, F.M.; Miller, J.B. Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Ann. Clin. Transl. Neurol. 2015, 2, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Ansseau, E.; Laoudj-Chenivesse, D.; Marcowycz, A.; Tassin, A.; Vanderplanck, C.; Sauvage, S.; Barro, M.; Mahieu, I.; Leroy, A.; Leclercq, I.; et al. DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS ONE 2009, 4, e7482. [Google Scholar] [CrossRef] [PubMed]
- Ansseau, E.; Eidahl, J.O.; Lancelot, C.; Tassin, A.; Matteotti, C.; Yip, C.; Liu, J.; Leroy, B.; Hubeau, C.; Gerbaux, C.; et al. Homologous Transcription Factors DUX4 and DUX4c Associate with Cytoplasmic Proteins during Muscle Differentiation. PLoS ONE 2016, 11, e0146893. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Facioscapulohumeral muscular dystrophy as a model for epigenetic regulation and disease. Antioxid. Redox Signal. 2015, 22, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2015, 24, R17–R23. [Google Scholar] [CrossRef] [PubMed]
- Daxinger, L.; Tapscott, S.J.; van der Maarel, S.M. Genetic and epigenetic contributors to FSHD. Curr. Opin. Genet. Dev. 2015, 33, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, M.-C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014, 83, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.I.; Yan, C.; Sapp, P.C.; McKenna-Yasek, D.; Kang, P.B.; Quinn, C.; Salameh, J.S.; King, O.D.; Jones, P.L. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenet. 2014, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; Goeman, J.J.; van der Vliet, P.J.; van Nieuwenhuizen, M.P.; Balog, J.; Vos-Versteeg, M.; Camano, P.; Ramos Arroyo, M.A.; Jerico, I.; Rogers, M.T.; et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015, 24, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Rost, S.; El Hajj, N.; Ferbert, A.; Deschauer, M.; Walter, M.C.; Schoser, B.; Tacik, P.; Kress, W.; Müller, C.R. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 2015, 23, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Lemmers, R.J.L.F.; Balog, J.; van der Vliet, P.J.; Lahaut, P.; van Nieuwenhuizen, M.P.; Straasheijm, K.R.; Debipersad, R.D.; Vos-Versteeg, M.; Salviati, L.; et al. The FSHD2 Gene SMCHD1 Is a Modifier of Disease Severity in Families Affected by FSHD1. Am. J. Hum. Genet. 2013, 93, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Stadler, G.; Rahimov, F.; King, O.D.; Chen, J.C.J.; Robin, J.D.; Wagner, K.R.; Shay, J.W.; Emerson, C.P.; Wright, W.E. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat. Struct. Mol. Biol. 2013, 20, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Attarian, S.; Salort-Campana, E.; Nguyen, K.; Behin, A.; Andoni Urtizberea, J. Recommendations for the management of facioscapulohumeral muscular dystrophy in 2011. Rev. Neurol. 2012, 168, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M.; Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology; Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 85, 357–364. [Google Scholar] [PubMed]
- Voet, N.; Bleijenberg, G.; Hendriks, J.; de Groot, I.; Padberg, G.; van Engelen, B.; Geurts, A. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in FSHD: An RCT. Neurology 2014, 83, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Laoudj-Chenivesse, D.; Carnac, G.; Bisbal, C.; Hugon, G.; Bouillot, S.; Desnuelle, C.; Vassetzky, Y.; Fernandez, A. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J. Mol. Med. 2005, 83, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.-C.; Böcker, K.; Hugon, G.; Pincemail, J.; et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Liu, J.; Domire, J.S.; Garwick-Coppens, S.E.; Guckes, S.M.; Mendell, J.R.; Flanigan, K.M.; Harper, S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: Implications for a targeted FSHD therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-W.; Snider, L.; Yao, Z.; Tawil, R.; Van Der Maarel, S.M.; Rigo, F.; Bennett, C.F.; Filippova, G.N.; Tapscott, S.J. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum. Mol. Genet. 2015, 24, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Scalpel or Straitjacket: CRISPR/Cas9 Approaches for muscular dystrophies. Trends Pharmacol. Sci. 2016, 37, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Choi, S.H.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet. Muscle 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Adams, A.M.; Johnsen, R.D.; Greer, K.; Moulton, H.M.; Wilton, S.D. Dystrophin isoform induction in vivo by antisense-mediated alternative splicing. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Takeshima, Y.; Nishio, H. Contributions of Japanese patients to development of antisense therapy for DMD. Brain Dev. 2016, 38, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Niks, E.H.; Aartsma-Rus, A. Exon skipping: A first in class strategy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2017, 17, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Carnac, G.; Flavier, S.; Mercier, J.; Vassetzky, Y.; Laoudj-Chenivesse, D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell. Mol. Med. 2010, 14, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.; Leroy, B.; Laoudj-Chenivesse, D.; Wauters, A.; Vanderplanck, C.; Le Bihan, M.-C.; Coppée, F.; Wattiez, R.; Belayew, A. FSHD Myotubes with Different Phenotypes Exhibit Distinct Proteomes. PLoS ONE 2012, 7, e51865. [Google Scholar] [CrossRef] [PubMed]
- Krom, Y.D.; Dumonceaux, J.; Mamchaoui, K.; den Hamer, B.; Mariot, V.; Negroni, E.; Geng, L.N.; Martin, N.; Tawil, R.; Tapscott, S.J.; et al. Generation of Isogenic D4Z4 Contracted and Noncontracted Immortal Muscle Cell Clones from a Mosaic Patient. Am. J. Pathol. 2012, 181, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Stern-Straeter, J.; Bonaterra, G.A.; Hörmann, K.; Kinscherf, R.; Goessler, U.R. Identification of valid reference genes during the differentiation of human myoblasts. BMC Mol. Biol. 2009, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- De la Kethulle de Ryhove, L.; Ansseau, E.; Nachtegael, C.; Pieters, K.; Vanderplanck, C.; Geens, M.; Sermon, K.; Wilton, S.D.; Coppée, F.; Lagneaux, L.; et al. The Role of D4Z4-Encoded Proteins in the Osteogenic Differentiation of Mesenchymal Stromal Cells Isolated from Bone Marrow. Stem Cells Dev. 2015, 24, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Marsollier, A.-C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum. Mol. Genet. 2016, 25, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Means to an end: Mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev. RNA 2014, 5, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. 3′ end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008, 27, 482–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Schmidt, R.; Gruber, A.R.; Martin, G.; Ghosh, S.; Belmadani, M.; Keller, W.; Zavolan, M. A comprehensive analysis of 3’ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016, 26, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Kapranov, P.; Foissac, S.; Kim, S.W.; Fishilevich, E.; Monaghan, A.P.; John, B.; Milos, P.M. Comprehensive Polyadenylation Site Maps in Yeast and Human Reveal Pervasive Alternative Polyadenylation. Cell 2010, 143, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3’ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Johnson, E.L.; Coller, H.A. Alternative polyadenylation can regulate post-translational membrane localization. Trends Cell Mol. Biol. 2015, 10, 37–47. [Google Scholar] [PubMed]
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.-W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Vanderplanck, C.; University of Mons, Mons, Belgium. Induction of double homeodomain protein DUX4c contributes to FSHD muscular dystrophy by interfering with myofibrillogenesis and myonuclear positioning. Unpublished work. 2017. [Google Scholar]
- Lek, A.; Rahimov, F.; Jones, P.L.; Kunkel, L.M. Emerging preclinical animal models for FSHD. Trends Mol. Med. 2015, 21, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Lee, Y.-C.; Yokota, T.; Chen, Y.-W. Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol. Ther. 2014, 22, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Ansseau, E.; Domire, J.S.; Wallace, L.M.; Eidahl, J.O.; Guckes, S.M.; Giesige, C.R.; Pyne, N.K.; Belayew, A.; Harper, S.Q. Aberrant splicing in transgenes containing introns, exons, and V5 epitopes: Lessons from developing an FSHD mouse model expressing a D4Z4 repeat with flanking genomic sequences. PLoS ONE 2015, 10, e0118813. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Bellgard, M.I.; Price, L.; Akkari, A.P.; Wilton, S.D. Translational development of splice-modifying antisense oligomers. Expert Opin. Biol. Ther. 2017, 17, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Ness, K.P.V.; Weller, D.L.; Poage, D.; Nelson, K.; Shrewsbury, A.S.B. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int. J. Toxicol. 2011, 30, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Carver, M.P.; Charleston, J.S.; Shanks, C.; Zhang, J.; Mense, M.; Sharma, A.K.; Kaur, H.; Sazani, P. Toxicological Characterization of exon skipping phosphorodiamidate morpholino oligomers (PMOs) in non-human primates. J. Neuromuscul. Dis. 2016, 3, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.A.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- O′Donovan, L.; Okamoto, I.; Arzumanov, A.A.; Williams, D.L.; Deuss, P.; Gait, M.J. Parallel synthesis of cell-penetrating peptide conjugates of PMO toward exon skipping enhancement in Duchenne muscular dystrophy. Nucleic Acid Ther. 2015, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansseau, E.; Vanderplanck, C.; Wauters, A.; Harper, S.Q.; Coppée, F.; Belayew, A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes 2017, 8, 93. https://doi.org/10.3390/genes8030093
Ansseau E, Vanderplanck C, Wauters A, Harper SQ, Coppée F, Belayew A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes. 2017; 8(3):93. https://doi.org/10.3390/genes8030093
Chicago/Turabian StyleAnsseau, Eugénie, Céline Vanderplanck, Armelle Wauters, Scott Q. Harper, Frédérique Coppée, and Alexandra Belayew. 2017. "Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD)" Genes 8, no. 3: 93. https://doi.org/10.3390/genes8030093
APA StyleAnsseau, E., Vanderplanck, C., Wauters, A., Harper, S. Q., Coppée, F., & Belayew, A. (2017). Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes, 8(3), 93. https://doi.org/10.3390/genes8030093