Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Clustering and Multiple Sequence Alignment
2.3. Evolutionary Analysis
2.4. Structural Prediction
2.5. Gene Ontology
2.6. Statistical Analysis
3. Results
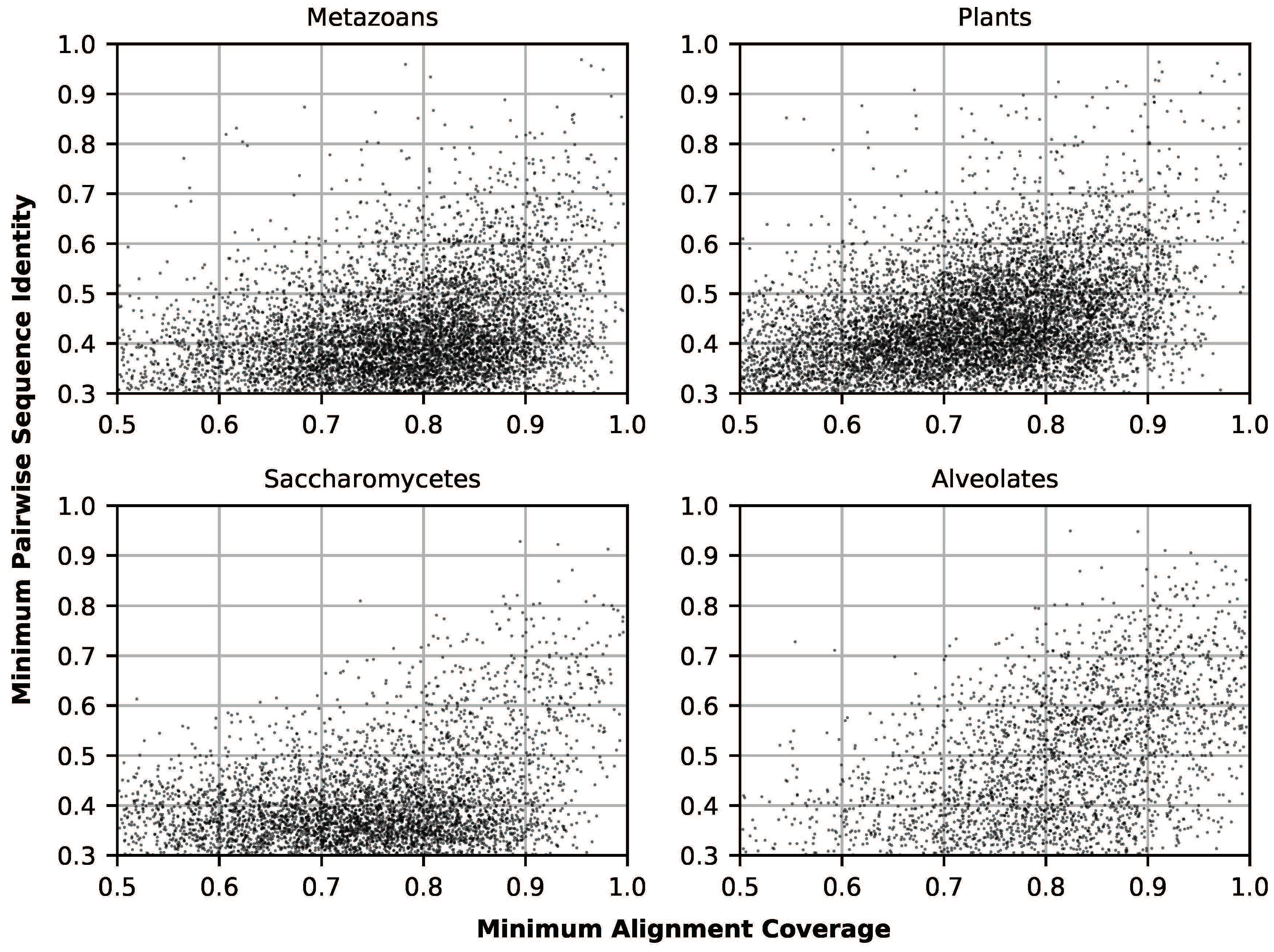
3.1. Clustering and Phylogenetics
3.2. Structural Prediction
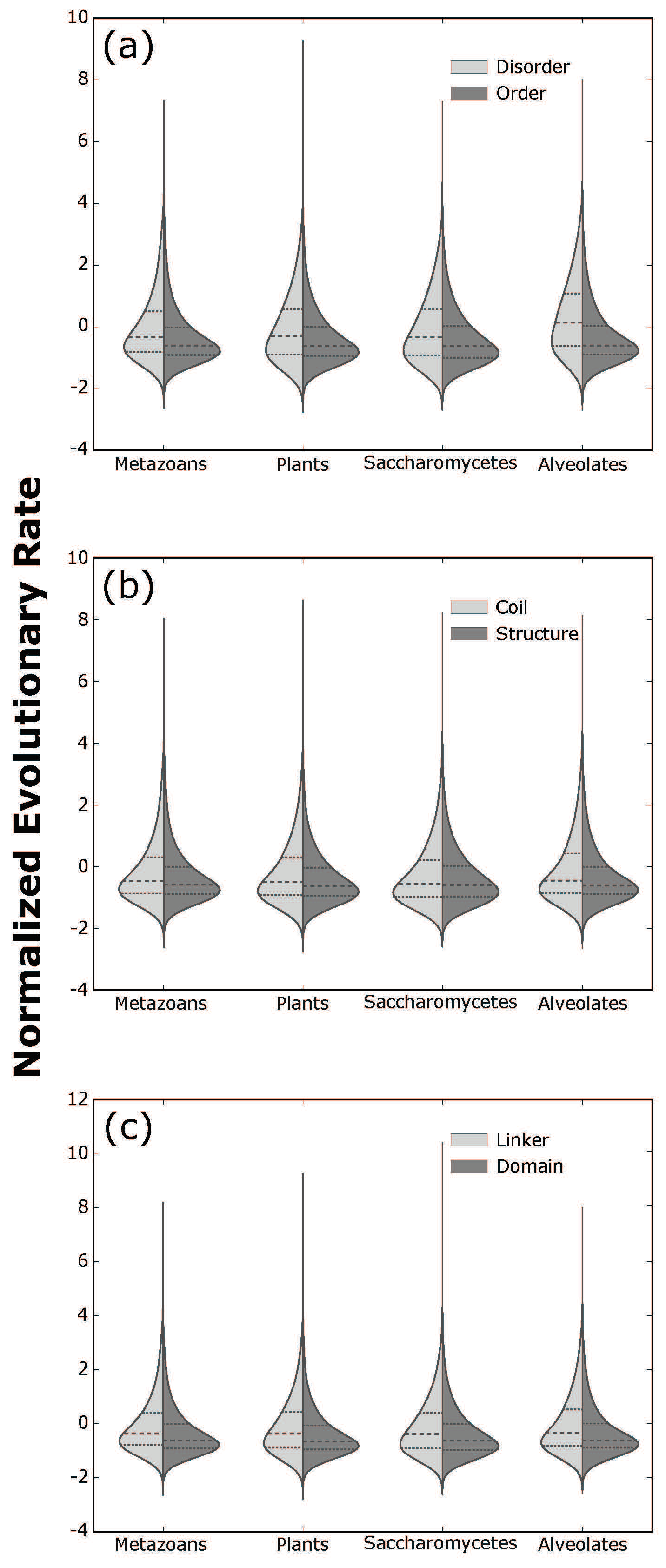
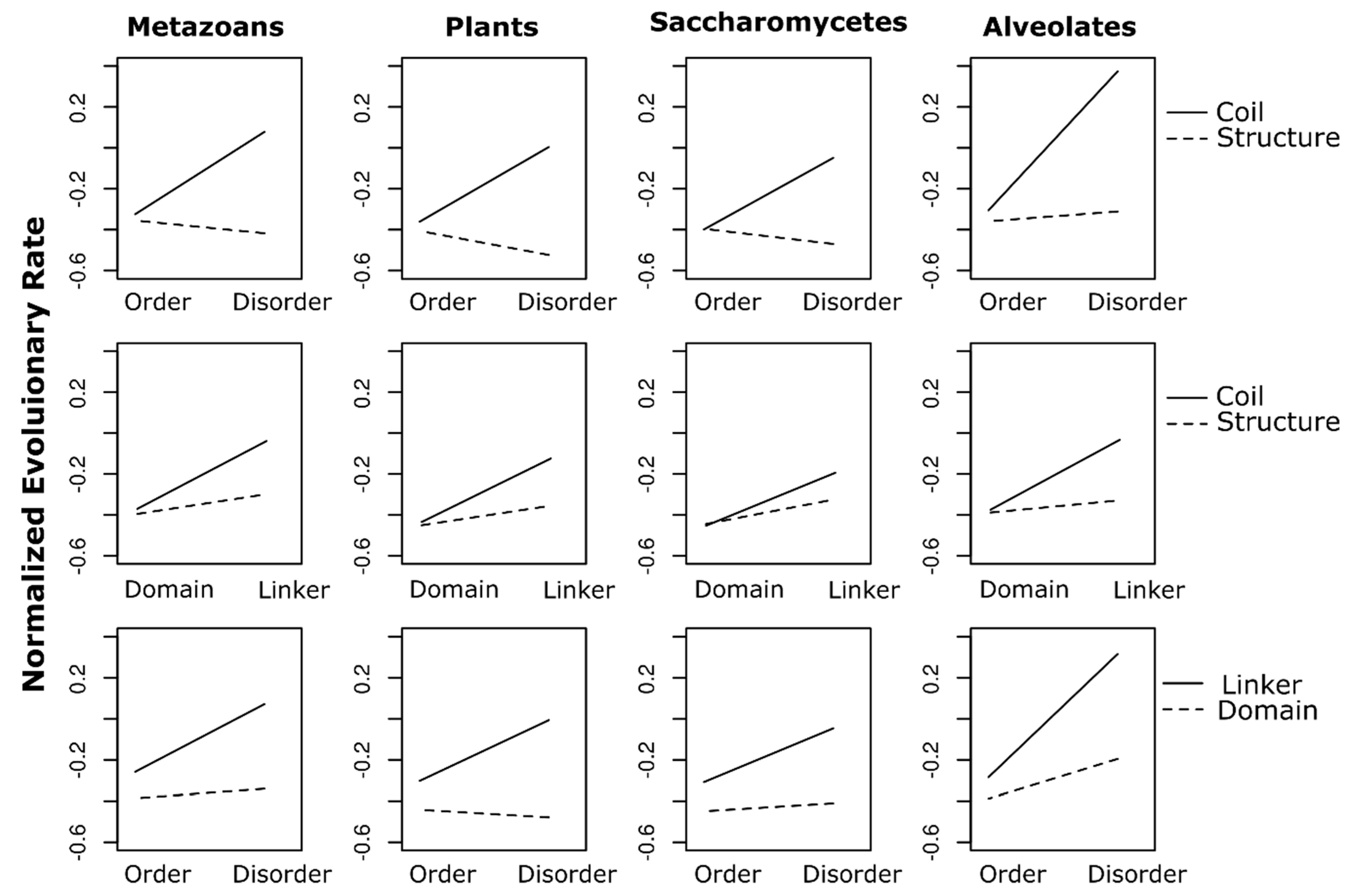
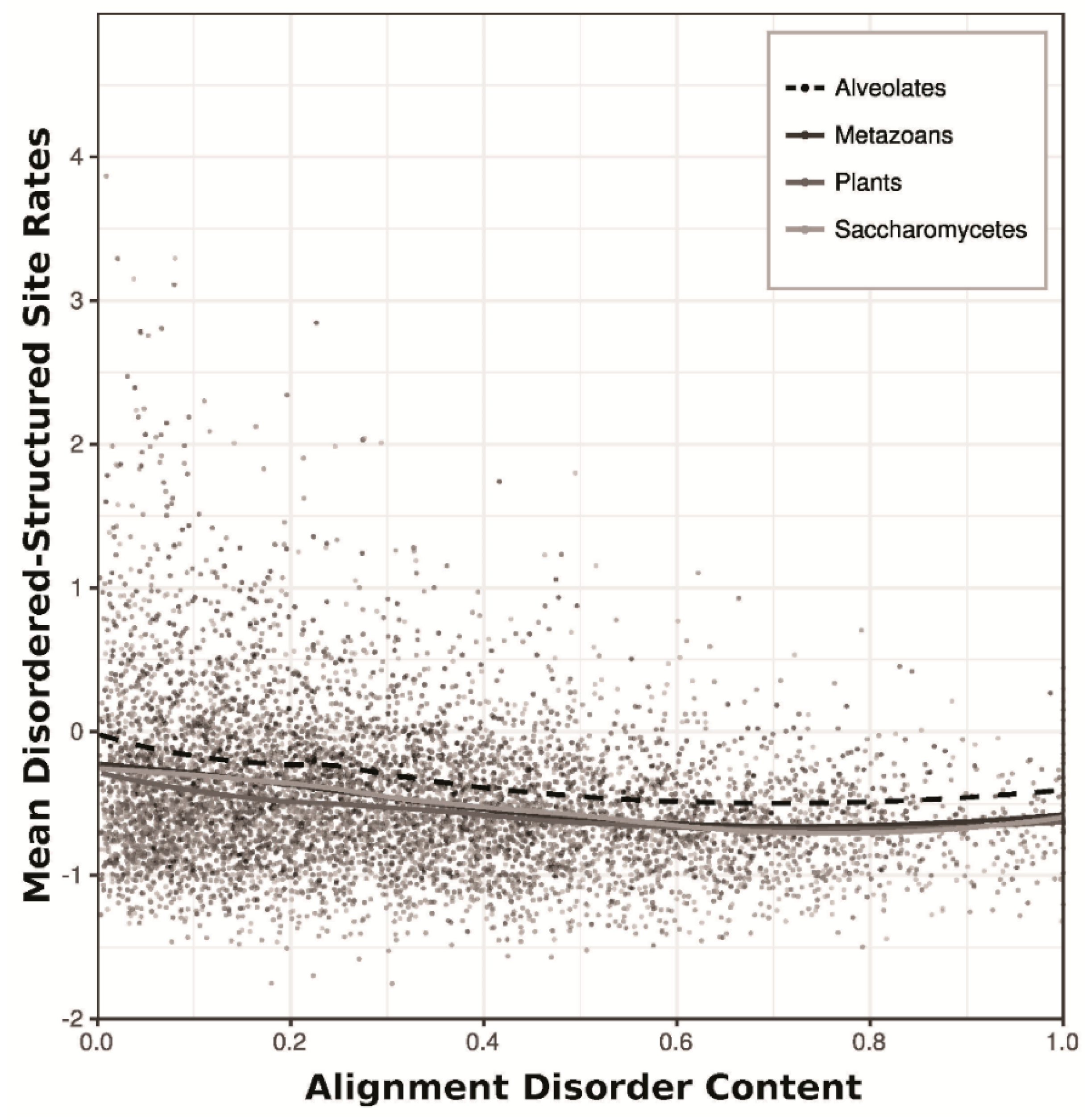
3.3. Statistical Analysis
3.4. Gene Ontology of Proteins with Disordered-Structured Sites
4. Discussion
4.1. Clustering and Phylogenetics
4.2. Structural Prediction
4.3. Gene Ontology
4.4. Intrinsic Disorder in Alveolates
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Kumar, S. Approximate methods for estimating the pattern of nucleotide substitution and the variation of substitution rates among sites. Mol. Biol. Evol. 1996, 13, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Echave, J.; Spielman, S.J.; Wilke, C.O. Causes of evolutionary rate variation among protein sites. Nat. Rev. Genet. 2016, 17, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Xia, Y. Structural determinants of protein evolution are context-sensitive at the residue level. Mol. Biol. Evol. 2009, 26, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-W.; Huang, T.-T.; Liu, J.-W.; Yu, S.-H.; Shih, C.-H.; Hwang, J.-K.; Echave, J. Local packing density is the main structural determinant of the rate of protein sequence evolution at site level. BioMed Res. Int. 2014, 2014, 572409. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Kendrew, J.C.; Watson, H.C. Structure and function of haemoglobin. J. Mol. Biol. 1965, 13, 669–678. [Google Scholar] [CrossRef]
- Kimura, M.; Ohta, T. On some principles governing molecular evolution. Proc. Natl. Acad. Sci. USA 1974, 71, 2848–2852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.-R. Determinants of the rate of protein sequence evolution. Nat. Rev. Genet. 2015, 16, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, J.; Dos Santos, H.G.; Siltberg-Liberles, J. The nuanced interplay of intrinsic disorder and other structural properties driving protein evolution. Mol. Biol. Evol. 2016, 33, 2248–2256. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Mayrose, I.; Graur, D.; Ben-Tal, N.; Pupko, T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol. Biol. Evol. 2004, 21, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Spielman, S.J.; Kosakovsky Pond, S.L.; Yeager, M. Relative evolutionary rates in proteins are largely insensitive to the substitution model. Mol. Biol. Evol. 2018, 35, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Sydykova, D.K.; Wilke, C.O. Theory of measurement for site-specific evolutionary rates in amino-acid sequences. bioRxiv 2018, 411025. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically disordered proteins with extensive annotations and literature. Nucleic Acids Res. 2012, 40, D507–D511. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, T.; Walsh, I.; Tosatto, S.C. Analysis and consensus of currently available intrinsic protein disorder annotation sources in the MobiDB database. BMC Bioinform. 2013, 14, S3. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. CDF it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Ward, S.M.; Lobley, A.E.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010, 38, W563–W568. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2011; Volume 1, p. 409. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Rossum, G. Python Reference Manual; Centrum voor Wiskunde en Informatica (CWI): Amsterdam, The Netherlands, 1995. [Google Scholar]
- Siegel, S.; Castellan, N.J. Nonparametric Statistics for the Behavioral Sciences; McGraw-Hill: New York, NY, USA, 1988; ISBN 0070573573. [Google Scholar]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; Sage: Thousand Oaks, CA, USA, 2011. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 9780387981413. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P.; Teslenko, M. MrBayes Version 3.2 Manual: Tutorials and Model Summaries. 2011. Available online: mrbayes.sourceforge.net/mb3.2_manual.pdf (accessed on 19 October 2018).
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J. Proteome Res. 2006, 5, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J. Roles of intrinsic disorder in protein-nucleic acid interactions. Mol. Biosyst. 2012, 8, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Zsolyomi, F.; Guharoy, M.; Tompa, P. Functional advantages of conserved intrinsic disorder in RNA-Binding proteins. PLoS ONE 2015, 10, e0139731. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, S.; Galardy, P.J.; Meester, W.J.N.; Ovaa, H.; Ploegh, H.L.; Gaudet, R. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J. Biol. Chem. 2005, 280, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.H.; Shoemaker, B.A.; Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V.; Panchenko, A.R. Intrinsic disorder in protein interactions: Insights from a comprehensive structural analysis. PLoS Comput. Biol. 2009, 5, e1000316. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Bellay, J.; Han, S.; Michaut, M.; Kim, T.; Costanzo, M.; Andrews, B.J.; Boone, C.; Bader, G.D.; Myers, C.L.; Kim, P.M. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011, 12, R14. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-P.; Zhang, X.; Han, P.; Arora, N.; Anders, R.F.; Norton, R.S. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol. Biochem. Parasitol. 2006, 150, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Guy, A.J.; Irani, V.; MacRaild, C.A.; Anders, R.F.; Norton, R.S.; Beeson, J.G.; Richards, J.S.; Ramsland, P.A. Insights into the immunological properties of intrinsically disordered malaria proteins using proteome scale predictions. PLoS ONE 2015, 10, e0141729. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.; Coetzer, T.L.; Blackledge, M.; Haertlein, M.; Mitchell, E.P.; Forsyth, V.T.; Jensen, M.R. Intrinsic disorder within the erythrocyte binding-like proteins from Plasmodium falciparum. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, A.; Bockwoldt, M.; Cooney, C.R.; Heiland, I.; Gossmann, T.I. Human long intrinsically disordered protein regions are frequent targets of positive selection. Genome Res. 2018, 28, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Siltberg-Liberles, J.; Grahnen, J.A.; Liberles, D.A. The evolution of protein structures and structural ensembles under functional constraint. Genes 2011, 2, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-W.; Liu, J.-W.; Yu, S.-H.; Shih, C.-H.; Hwang, J.-K.; Echave, J. Site-specific structural constraints on protein sequence evolutionary divergence: Local packing density versus solvent exposure. Mol. Biol. Evol. 2014, 31, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Metazoans | Plants | Saccharomycetes | Alveolates |
|---|---|---|---|---|
| Clusters | 6938 | 8266 | 4494 | 2697 |
| Sequences | 130632 | 198081 | 122132 | 44060 |
| Total Alignment Sites | 4677490 | 4703587 | 2990109 | 1640297 |
| Gap-free sites | 3217225 | 2851827 | 1954761 | 1179122 |
| Ordered Sites | 1819695 | 1706275 | 1223656 | 801629 |
| Disordered Sites | 373639 | 234853 | 125047 | 113892 |
| Structured sites | 1062380 | 1014001 | 722444 | 417702 |
| Random coil sites | 1314563 | 1064725 | 670357 | 424795 |
| Domain sites | 1436746 | 1175745 | 936813 | 422813 |
| Linker sites | 1368702 | 1289830 | 817371 | 657080 |
| Median Order Rate | −0.599 | −0.625 | −0.6188 | −0.605 |
| Median Disorder Rate | −0.3155 | −0.2916 | −0.3271 | 0.1426 |
| Median Structure Rate | −0.5787 | −0.6262 | −0.5935 | −0.605 |
| Median Coil Rate | −0.4682 | −0.5013 | −0.5603 | −0.4542 |
| Median Domain Rate | −0.62345 | −0.6679 | −0.6353 | −0.629 |
| Median Linker Rate | −0.3698 | −0.3718 | −0.3902 | −0.3569 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahrens, J.B.; Rahaman, J.; Siltberg-Liberles, J. Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains. Genes 2018, 9, 553. https://doi.org/10.3390/genes9110553
Ahrens JB, Rahaman J, Siltberg-Liberles J. Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains. Genes. 2018; 9(11):553. https://doi.org/10.3390/genes9110553
Chicago/Turabian StyleAhrens, Joseph B., Jordon Rahaman, and Jessica Siltberg-Liberles. 2018. "Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains" Genes 9, no. 11: 553. https://doi.org/10.3390/genes9110553
APA StyleAhrens, J. B., Rahaman, J., & Siltberg-Liberles, J. (2018). Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains. Genes, 9(11), 553. https://doi.org/10.3390/genes9110553