Renal Tubule Repair: Is Wnt/?-Catenin a Friend or Foe?

Abstract
:1. Introduction
2. Wnt/β-Catenin Signaling in Kidney Development
3. Wnt/β-Catenin Signaling in Epithelial Injury
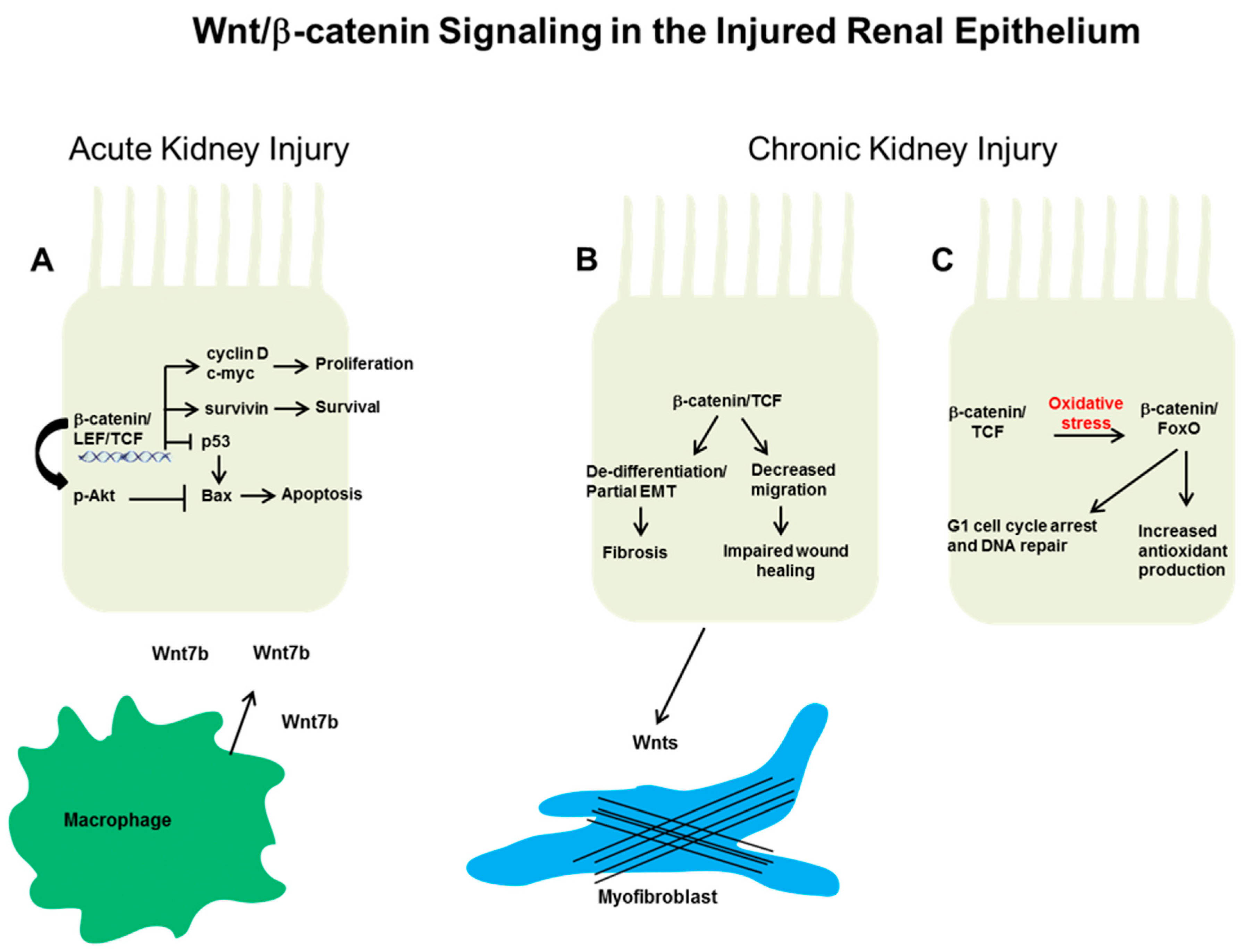
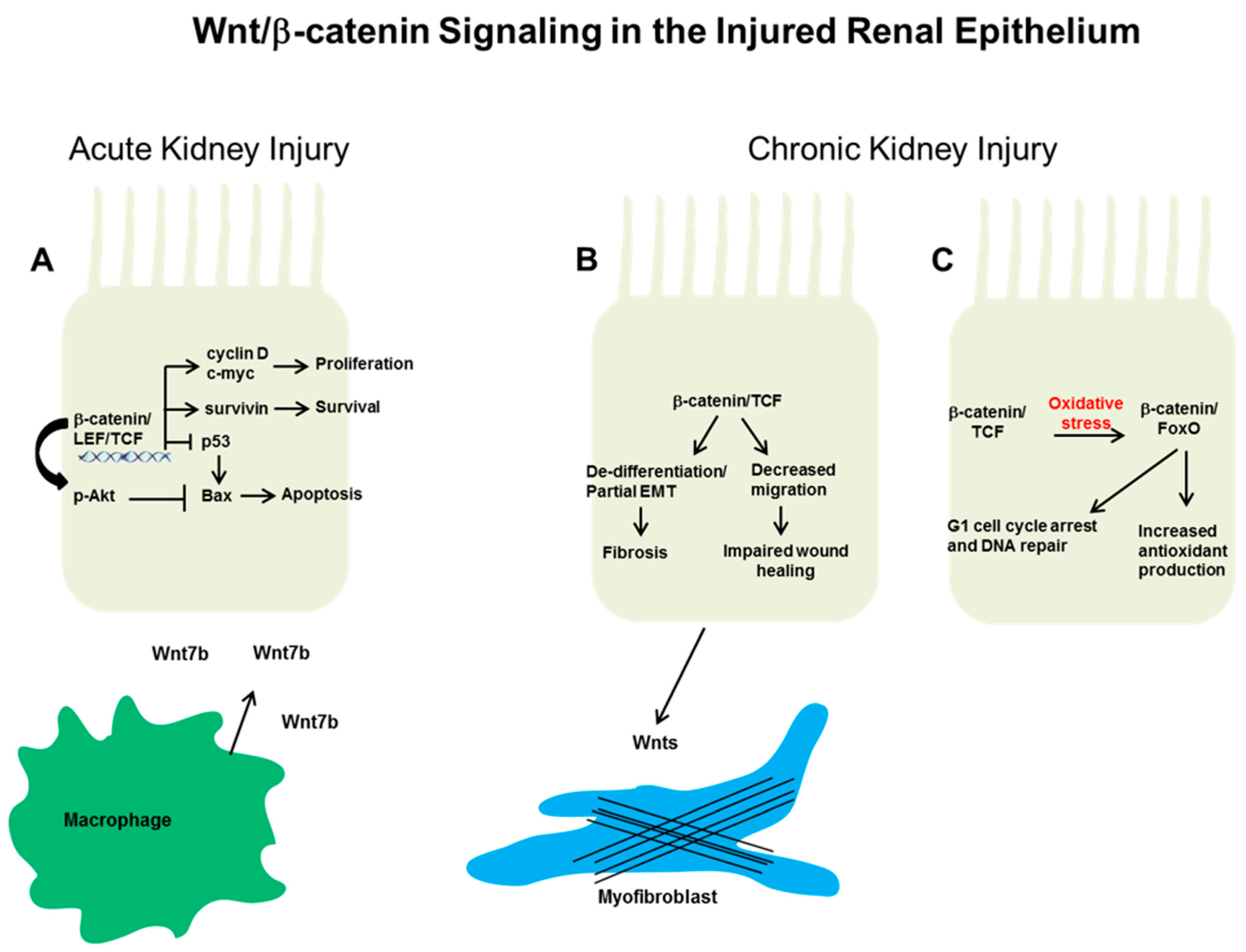
3.1. Acute Kidney Injury
3.2. Wnt/β-Catenin Signaling in Tubular Progenitors after Injury
4. Wnt/β-Catenin Epithelial Signaling in Chronic Kidney Disease
4.1. Systemic Inhibitors of Wnt/β-Catenin
4.2. Epithelial Wnt/β-Catenin and Chronic Kidney Disease
5. Therapeutic Implications and Future Directions
Acknowledgments
Conflicts of Interest
References
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003, 348, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Humphreys, B.D.; Bonventre, J.V. Pathophysiology of acute kidney injury to chronic kidney disease: Maladaptive repair. Contrib. Nephrol. 2011, 174, 149–155. [Google Scholar] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Boil. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Goggolidou, P. Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis 2014, 10, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, D.; Cox, B.; Cain, J.; Lau, A.; Athaide, V.; Gill, P.S.; Kuure, S.; Sainio, K.; Rosenblum, N.D. Canonical Wnt/β-catenin signaling is required for ureteric branching. Dev. Boil. 2008, 317, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Maretto, S.; Cordenonsi, M.; Dupont, S.; Braghetta, P.; Broccoli, V.; Hassan, A.B.; Volpin, D.; Bressan, G.M.; Piccolo, S. Mapping Wnt/β-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 3299–3304. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Vaquer, A.; Piliszek, A.; Tian, G.; Aho, R.J.; Dufort, D.; Hadjantonakis, A.K. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev. Biol. 2010, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ren, S.; Duffield, J.S. Wnt signalling in kidney diseases: Dual roles in renal injury and repair. J. Pathol. 2013, 229, 221–231. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/β-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.; Eder, S.; Perco, P.; Enrich, J.; Scheiber, K.; Koppelstätter, C.; Schratzberger, G.; Mayer, B.; Oberbauer, R.; Meyer, T.W.; et al. Gene expression profiles of human proximal tubular epithelial cells in proteinuric nephropathies. Kidney Int. 2007, 71, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.B. Growth and death in the developing mammalian kidney: Signals, receptors and conversations. Bioessays 2002, 24, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.M.; Hueber, P.A.; Chu, L.; Campbell, R.; Patenaude, A.M.; Dziarmaga, A.J.; Quinlan, J.; Mohamed, O.; Dufort, D.; Goodyer, P.R. Canonical Wnt signaling during kidney development. Am. J. Physiol. Ren. Physiol. 2007, 293, F494–F500. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.J.; Park, J.S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kispert, A.; Vainio, S.; McMahon, A.P. Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 1998, 125, 4225–4234. [Google Scholar] [PubMed]
- Park, J.S.; Valerius, M.T.; McMahon, A.P. Wnt/β-catenin signaling regulates nephron induction during mouse kidney development. Development 2007, 134, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 1994, 372, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Wang, H.; Yang, Y.; Sharma, N.; Tarasova, N.; Ajima, R.; Yamaguchi, T.P.; Rodriguez, L.G.; Perantoni, A.O. Wnt4 induces nephronic tubules in metanephric mesenchyme by a non-canonical mechanism. Dev. Boil. 2011, 352, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Marose, T.D.; Merkel, C.E.; McMahon, A.P.; Carroll, T.J. β-catenin is necessary to keep cells of ureteric bud/Wolffian duct epithelium in a precursor state. Dev. Boil. 2008, 314, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ott, K.M.; Barasch, J. Wnt/β-catenin signaling in nephron progenitors and their epithelial progeny. Kidney Int. 2008, 74, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.; Boivin, F.; Li, A.; Lim, J.; Svajger, B.; Rosenblum, N.D.; Bridgewater, D. β-Catenin overexpression in the metanephric mesenchyme leads to renal dysplasia genesis via cell-autonomous and non-cell-autonomous mechanisms. Am. J. Pathol. 2014, 184, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, D.; Di Giovanni, V.; Cain, J.E.; Cox, B.; Jakobson, M.; Sainio, K.; Rosenblum, N.D. β-catenin causes renal dysplasia via upregulation of TGFβ2 and DKK1. J. Am. Soc. Nephrol. 2011, 22, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ma, W.; O’Brien, L.L.; Chung, E.; Guo, J.J.; Cheng, J.G.; Valerius, M.T.; McMahon, J.A.; Wong, W.H.; McMahon, A.P. SIX2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 2012, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Pietila, I.; Ellwanger, K.; Railo, A.; Jokela, T.; Barrantes Idel, B.; Shan, J.; Niehrs, C.; Vainio, S.J. Secreted Wnt antagonist Dickkopf-1 controls kidney papilla development coordinated by Wnt-7b signalling. Dev. Boil. 2011, 353, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Tanaka, H.; Okado, T.; Shimamura, H.; Inoshita, S.; Kuwahara, M.; Sasaki, S. Expression and function of the developmental gene Wnt-4 during experimental acute renal failure in rats. J. Am. Soc. Nephrol. 2003, 14, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.P.; Kobayashi, A.; Taketo, M.M.; McMahon, A.P.; Humphreys, B.D. Wnt4/β-catenin signaling in medullary kidney myofibroblasts. J. Am. Soc. Nephrol. 2013, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Li, B.; Rao, S.; Yeo, E.J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Li, Y.; Lin, L.; Zhou, L.; Igarashi, P.; Liu, Y. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int. 2012, 82, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Havasi, A.; Gall, J.M.; Mao, H.; Schwartz, J.H.; Borkan, S.C. β-catenin promotes survival of renal epithelial cells by inhibiting BAX. J. Am. Soc. Nephrol. 2009, 20, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.; Tao, S.; Yang, H.C.; Fogo, A.B.; Woodgett, J.R.; Harris, R.C.; Rao, R. Specific deletion of glycogen synthase kinase-3β in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int. 2012, 82, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Havasi, A.; Gall, J.; Bonegio, R.; Li, Z.; Mao, H.; Schwartz, J.H.; Borkan, S.C. GSK3β promotes apoptosis after renal ischemic injury. J. Am. Soc. Nephrol. 2010, 21, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Wang, Z.; Zhuang, S.; Dworkin, L.; Peng, A.; Gong, R. Delayed administration of a single dose of lithium promotes recovery from AKI. J. Am. Soc. Nephrol. 2014, 25, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, O.H.; Aravamudhan, A.; Rangarajan, D.; Kusaba, T.; Zhang, V.; Welborn, J.; Gauvin, D.; Hou, X.; Kramann, R.; Humphreys, B.D. Paracrine Wnt1 Drives Interstitial Fibrosis without Inflammation by Tubulointerstitial Cross-Talk. J. Am. Soc. Nephrol. 2016, 27, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Tan, R.J.; Zhou, L.; Li, Y.; Liu, Y. Kidney tubular β-catenin signaling controls interstitial fibroblast fate via epithelial-mesenchymal communication. Sci. Rep. 2013, 3, 1878. [Google Scholar] [CrossRef] [PubMed]
- Nlandu-Khodo, S.; Neelisetty, S.; Phillips, M.; Manolopoulou, M.; Bhave, G.; May, L.; Clark, P.E.; Yang, H.; Fogo, A.B.; Harris, R.C.; et al. Blocking TGF-β and β-Catenin Epithelial Crosstalk Exacerbates CKD. J. Am. Soc. Nephrol. 2017, 28, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.Q.; Freedman, B.S.; Morizane, R.; Lerou, P.H.; Valerius, M.T.; Bonventre, J.V. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 2014, 25, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Hendry, C.E.; Vanslambrouck, J.M.; Ineson, J.; Suhaimi, N.; Takasato, M.; Rae, F.; Little, M.H. Direct transcriptional reprogramming of adult cells to embryonic nephron progenitors. J. Am. Soc. Nephrol. 2013, 24, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Montoro, D.T.; Contreras-Trujillo, H.; Harari-Steinberg, O.; Newman, A.M.; Tsai, J.M.; Lim, X.; Van-Amerongen, R.; Bowman, A.; Januszyk, M.; et al. In vivo clonal analysis reveals lineage-restricted progenitor characteristics in mammalian kidney development, maintenance, and regeneration. Cell Rep. 2014, 7, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Humphreys, B.D. Report on ISN Forefronts, Florence, Italy, 12–15 September 2013: Stem cells and kidney regeneration. Kidney Int. 2014, 86, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Iglesias, D.M.; Corsini, R.; Chu, L.; Goodyer, P. Wnt/β-Catenin Signaling Is Required for Integration of CD24+ Renal Progenitor Cells into Glycerol-Damaged Adult Renal Tubules. Stem Cells Int. 2015, 2015, 391043. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J. Am. Soc. Nephrol. 2015, 26, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Patel, M.B.; Zhang, J.; Bunte, R.M.; Rudemiller, N.P.; Griffiths, R.; Virshup, D.M.; Crowley, S.D. Experimental inhibition of porcupine-mediated Wnt O-acylation attenuates kidney fibrosis. Kidney Int. 2016, 89, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhou, D.; Tan, R.J.; Fu, H.; Zhou, L.; Hou, F.F.; Liu, Y. Sustained Activation of Wnt/β-Catenin Signaling Drives AKI to CKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Surendran, K.; Schiavi, S.; Hruska, K.A. Wnt-dependent β-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J. Am. Soc. Nephrol. 2005, 16, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Fu, H.; Zhang, L.; Zhang, K.; Min, Y.; Xiao, L.; Lin, L.; Bastacky, S.I.; Liu, Y. Tubule-Derived Wnts Are Required for Fibroblast Activation and Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2322–2336. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Rao, P.; Zhang, Y.; Liu, L.; Pang, M.; Wang, H.; Hu, M.; Tian, X.; Zhang, J.; Zhao, Y.; et al. Redirecting TGF-β Signaling through the β-catenin/FOXO Complex Prevents Kidney Fibrosis. J. Am. Soc. Nephrol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [PubMed]
- Zemans, R.L.; Briones, N.; Campbell, M.; McClendon, J.; Young, S.K.; Suzuki, T.; Yang, I.V.; De Langhe, S.; Reynolds, S.D.; Mason, R.J.; et al. Neutrophil transmigration triggers repair of the lung epithelium via β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15990–15995. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; de Vries-Smits, L.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.; Korswagen, H.C. Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fergusson, M.M.; Wu, J.J.; Rovira, I.I.; Liu, J.; Gavrilova, O.; Lu, T.; Bao, J.; Han, D.; Sack, M.N.; et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011, 4, ra6. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yin, J.; Wang, H.; Jiang, G.; Deng, M.; Zhang, G.; Bu, X.; Cai, S.; Du, J.; He, Z. FOXO3a modulates Wnt/β-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells. Cell. Signal. 2015, 27, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; Almeida, M. Gone with the Wnts: β-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol. Endocrinol. 2007, 21, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Boil. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between snail and LEF-1 transcription factors is essential for TGF-β1-induced epithelial-mesenchymal transition. Mol. Boil. Cell 2006, 17, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of β-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Ramesh, G.; Sun, L.; Dong, Z. Impaired wound healing in hypoxic renal tubular cells: Roles of hypoxia-inducible factor-1 and glycogen synthase kinase 3β/β-catenin signaling. J. Pharmacol. Exp. Ther. 2012, 340, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Beaton, H.; Andrews, D.; Parsons, M.; Murphy, M.; Gaffney, A.; Kavanagh, D.; McKay, G.J.; Maxwell, A.P.; Taylor, C.T.; Cummins, E.P.; et al. Wnt6 regulates epithelial cell differentiation and is dysregulated in renal fibrosis. Am. J. Physiol. Renal. Physiol. 2016, 311, F35–F45. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell α3β1 integrin links β-catenin and SMAD signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhou, L.; Wang, Y.; Miao, J.; Hong, X.; Hou, F.F.; Liu, Y. (Pro)renin Receptor Is an Amplifier of Wnt/β-Catenin Signaling in Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2393–2408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Manipulation of Wnt/β-Catenin Pathway | Injury Model | Response | Mechanism | Ref. |
|---|---|---|---|---|
| Deletion of Wnt7 in macrophages | I/R | Increased renal tubular apoptosis | Cell cycle progression and basement membrane repair | [32] |
| Tubule specific ablation of β-catenin | I/R and folic acid | Greater mortality and tubular apoptosis, lower renal function | Increased pro-apototic Bax and p53, decreased pAkt and survivin | [33] |
| GSK-3β inhibition in proximal tubule | HgCl2 | Reduced tubular apoptosis and mortality, improved function | Increased cell proliferation and cyclin D1/c-Myc | [35] |
| Wnt1 overexpression by proximal tubule | None | Increased interstitial fibrosis | Increased paracrine myofibroblast signaling, no epithelial injury | [38] |
| Tubule specific ablation of β-catenin | UUO | No effect on fibrosis, reduced epithelial de-differentiation, increased fibroblast survival | Fibroblast survival due to reduced MMP-7-dependent FasL induction | [39] |
| Proximal tubule specific β-catenin stabilization (cells also lack TGF-β receptor) | Aristolochic acid | Reduced tubulointerstitial fibrosis, improved renal function, and reduced tubular injury | Reduced susceptibility to apoptosis and decreased G2/M arrest | [40] |
| Mutation of Frizzled4 receptor, primarily expressed in epithelia | I/R | Persistent epithelial injury | Increased apoptosis | [32] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gewin, L.S. Renal Tubule Repair: Is Wnt/?-Catenin a Friend or Foe? Genes 2018, 9, 58. https://doi.org/10.3390/genes9020058
Gewin LS. Renal Tubule Repair: Is Wnt/?-Catenin a Friend or Foe? Genes. 2018; 9(2):58. https://doi.org/10.3390/genes9020058
Chicago/Turabian StyleGewin, Leslie S. 2018. "Renal Tubule Repair: Is Wnt/?-Catenin a Friend or Foe?" Genes 9, no. 2: 58. https://doi.org/10.3390/genes9020058
APA StyleGewin, L. S. (2018). Renal Tubule Repair: Is Wnt/?-Catenin a Friend or Foe? Genes, 9(2), 58. https://doi.org/10.3390/genes9020058