Natural Formation of Chloro- and Bromoacetone in Salt Lakes of Western Australia

 , , and
, , and
Abstract
:1. Introduction
2. Experimental Section
3. Results
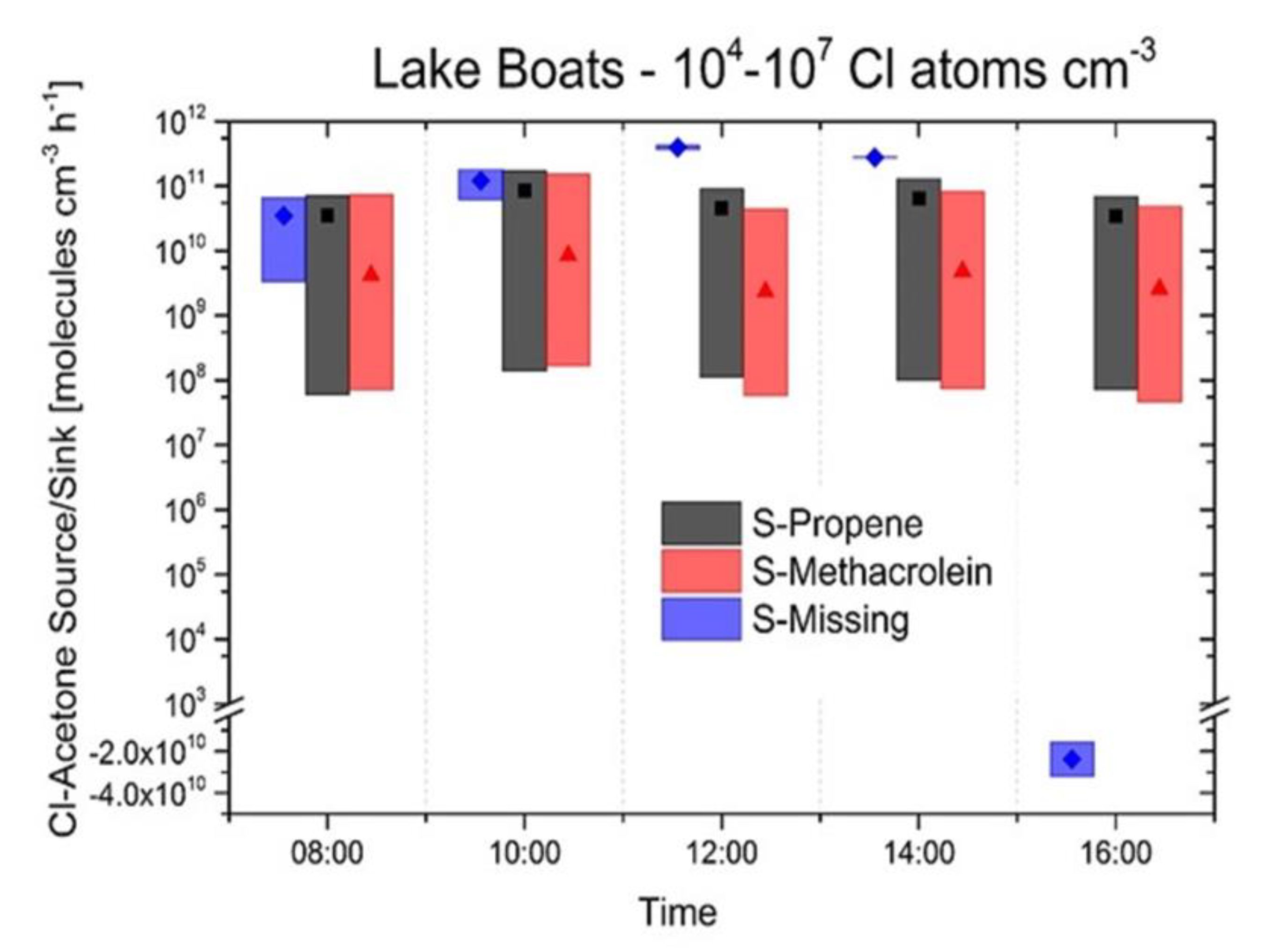
3.1. Lake Measurements
3.2. Source and Sink Balance Model
3.3. Humic Acid as a Source of Haloacetones
3.4. pH-Dependent Bromoacetone Formation
3.5. Hydrolysis and Nucleophilic Substitution of Bromoacetone
3.6. Photolysis Rates of Bromoacetone
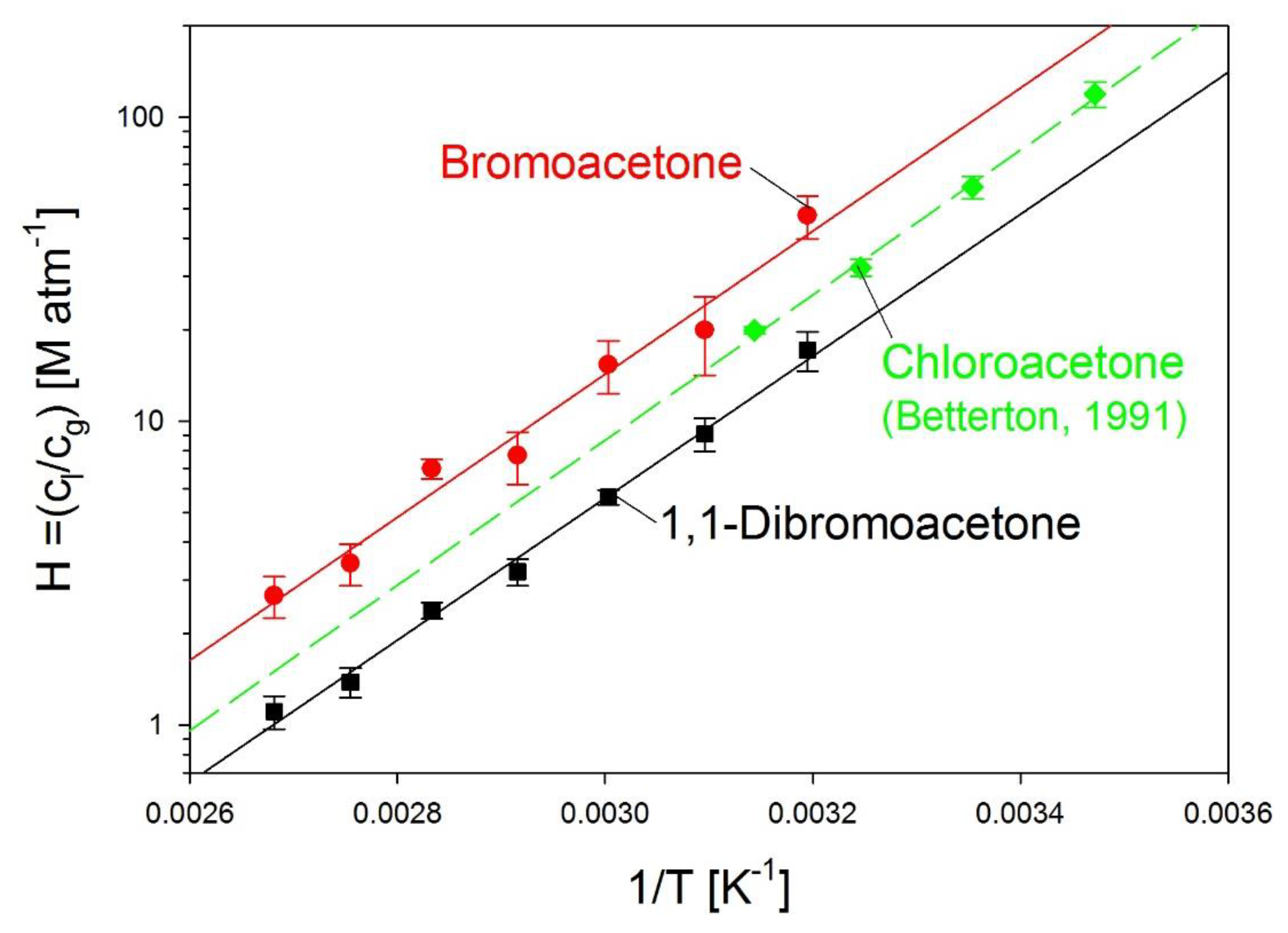
3.7. Henry’s Law Constants of Bromoacetone and 1,1-dibromoacetone
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gribble, G.W. Naturally Occurring Organohalogen Compounds—A Comprehensive Update; Springer: Wien, Austria, 2010. [Google Scholar]
- Schöler, H.F.; Keppler, F. Abiotic Formation of Organohalogens during Early Diagenetic Processes; The Handbook of Environmental Chemistry; Springer Verlag: Heidelberg, Germany, 2003; Volume 3. [Google Scholar]
- Gribble, G.W. The Diversity of Naturally Produced Organohalogens; Chemosphere: Heidelberg, Germany, 2003; Volume 52, pp. 289–297. [Google Scholar]
- Comba, P.; Kerscher, M.; Krause, T.; Schöler, H.F. Iron-catalysed oxidation and halogenation of organic matter in nature. Environ. Chem. 2015, 12, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Behnke, W.; Zetzsch, C. Efficiency of Br− and I− for activating chloride in sea-spray aerosol, derived from aerosol smog chamber experiments and model calculation. Geophys. Res. Abstr. 2005, 7, 03095. [Google Scholar]
- Schöler, H.F.; Zetzsch, C.; Kappler, A. Foreword to the Special Issue on ‘Naturally Produced Organohalogens—Atmosphere, Soil’. Environ. Chem. 2015, 12, 1–2. [Google Scholar] [CrossRef]
- Abrol, I.P.; Massoud, F.I. Salt-Affected Soils and Their Management 1988; Food and Agriculture Organization of the United Nations: Rome, Italy, 1988; pp. 10–12. [Google Scholar]
- Krause, T.; Tubbesing, C.; Benzing, K.; Schöler, H.F. Model reactions and natural occurence of furans from hypersaline environments. Biogeoscience 2013, 11, 2871–2882. [Google Scholar] [CrossRef]
- Krause, T. Natural Occurence of Volatile Mono-/Polyhalogenated and Aromatic/Heteroaromatic Hydrocarbons from Hypersaline Environments. Ph.D. Thesis, University of Heidelberg, Heidelberg, Germany, 2014. [Google Scholar]
- Mulder, I.; Krause, T.; Sattler, T.; Tubbesing, C.; Studenroth, S.; Bukowski, K.; Atlas, E.; Schöler, H.F. Thermolytic degradation of methyl methionine and implications for its role in DMS and MeCl formation in hypersaline environments. Environ. Chem. 2015, 12, 415. [Google Scholar] [CrossRef]
- Reckhow, D.A.; Singer, P.C. Chlorination by-products in drinking waters: From formation potentials to finished water concentrations. Am. Water Works Assoc. 1990, 82, 173–180. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, L.Y.; Peng, H.S. Effect of NOM characteristics on brominated organics formation by ozonation. Environ. Int. 2004, 29, 1049–1055. [Google Scholar] [CrossRef]
- McKague, A.B.; Bradley, D.; Meier, H.P.; Monteith, D.; Betts, J.L. Chloroacetones in pulp mill chlorination-stage effluents. Environ. Toxicol. Chem. 1990, 9, 1301–1303. [Google Scholar] [CrossRef]
- Rappe, C. Halogenation of ketones. Acta Chem. Scand. 1966, 20, 376–384. [Google Scholar] [CrossRef]
- Da Rosa, M.B.; Behnke, W.; Zetzsch, C. Study of the heterogeneous reaction of O3 with CH3SCH3 using the wetted-wall flowtube technique. Atmos. Chem. Phys. 2003, 3, 1665–1673. [Google Scholar] [CrossRef]
- Hughes, E.D.; Watson, H.B.; Yates, E.D. Constitutional factors controlling prototropic change in carbonyl compounds. Part I. The relative speeds of enolisation of acetone and bromoacetone and the effect of the acid catalyst. Catalyst. J. Chem. Soc. 1931, 6, 3318–3324. [Google Scholar] [CrossRef]
- Watson, B.; Yates, E.D. Constitutional factors controlling prototropic changes in carbonyl compounds. Part 2. Acid and base catalysis in the bromination of halogenated acetones. Catalyst. J. Chem. Soc. 1932, 7, 1207–1215. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Cossar, J. The chlorination of acetone: a complete kinetic analysis. Can. J. Chem. 1986, 64, 1250–1266. [Google Scholar] [CrossRef]
- Cox, R.A.; Warkentin, J. Kinetics of bromination of acetone, bromoacetone, and 1,1-dibromoacetone. Canandian J. Chem. 1970, 50, 3233–3238. [Google Scholar] [CrossRef]
- McKague, A.B.; Lee, E.G.H.; Douglas, G.R. Chloroacetones: Mutagenic constituents of bleached kraft chlorination effluents. Mutation Res. 1981, 91, 301–306. [Google Scholar] [CrossRef]
- Osterman-Golkar, S. Reaction kinetics in water of chloroethylene oxide, chloroacetaldehyde, and chloroacetone. Hereditas 1984, 101, 65–68. [Google Scholar] [CrossRef]
- Keil, A.D.; Shepson, P.B. Chlorine and bromine atom ratios in the springtime arctic troposphere as determined from measurements of halogenated volatile organic compounds. J. Geophys. Res. Atmos. 2006, 111, D17. [Google Scholar] [CrossRef]
- Kleindienst, T.E.; Shepson, P.B.; Nero, C.M.; Bufalini, J.J. The production of chlorine atom from the reaction of OH with chlorinated hydrocarbons. Int. J. Chem. Kinet. 1989, 21, 863–884. [Google Scholar] [CrossRef]
- Kaiser, E.W.; Wallington, T.J. Pressure dependence of the reaction Cl + C3H6. J. Phys. Chem. 1996, 100, 9788–9793. [Google Scholar] [CrossRef]
- Barnes, I.; Bastian, V.; Becker, K.H.; Overath, R.; Tong, Z. Rate constants for the reactions of Br atoms with a series of alkanes, alkenes, and alkynes in the presence of O2. Int. J. Chem. Kinet. 1989, 21, 499–517. [Google Scholar] [CrossRef]
- Wallington, T.J.; Skewes, L.M.; Siegl, W.O.; Japar, S.M. A relative rate study of the reaction of bromine atoms with a variety of organic compounds at 295 K. Int. J. Chem. Kinet. 1989, 21, 1069–1076. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S.; Apel, E.C.; Riemer, D.D.; Paulson, S.E. Rate coefficients and mechanisms of the reaction of Cl-atoms with a series of unsaturated hydrocarbons under atmospheric conditions. Int. J. Chem. Kinet. 2003, 35, 334–353. [Google Scholar] [CrossRef]
- Calvert, J.G.; Orlando, J.J.; Stockwell, W.R.; Wallington, T.J. The Mechanisms of Reactions Influencing Atmospheric Ozone; Oxford University Press: New York, NY, USA, 2015; pp. 67–96. [Google Scholar]
- Canosa-Mas, C.E.; Cotter, E.S.N.; Duffy, J.; Thompson, K.C.; Wayne, R.P. The reaction of atomic chlorine with acrolein, methacrolein and methyl vinyl ketone. Phys. Chem. Chem. Phys. 2001, 3, 3075–3084. [Google Scholar] [CrossRef]
- Sauer, C.G.; Barnes, I.; Becker, K.H. FT-IR kinetic and product study of the Br-radical initiated oxidation of α, β-unsaturated organic carbonyl compounds. Atmos. Environ. 1999, 33, 1969–2979. [Google Scholar] [CrossRef]
- Burkholder, J.B.; Gilles, M.K.; Gierczak, T.; Ravishankara, A.R. The atmospheric degradation of 1-bromopropane: The photochemistry of bromoacetone. Geophys. Res. Lett. 2002, 29, 1822. [Google Scholar] [CrossRef]
- Markert, F.; Nielsen, O.J. The reactions of OH radicals with chloroalkanes in the temperature range 295–360 K. Chem. Phys. Lett. 1992, 194, 123–127. [Google Scholar] [CrossRef]
- Donaghy, T.; Shanahan, I.; Hande, M.; Fitzpatrick, S. Rate constants and atmospheric lifetimes for the reactions of OH radicals and Cl atoms with haloalkanes. Int. J. Chem. Kinet. 1993, 25, 273–284. [Google Scholar] [CrossRef]
- Calvert, J.G.; Derwent, R.G.; Orlando, J.J.; Tyndall, G.S.; Wallington, T.J. Mechanisms of Atmospheric Oxidation of the Alkanes; Oxford University Press: New York, NY, USA, 2008; pp. 379–603. [Google Scholar]
- Nelson, D.D.; Wormhoudt, J.C.; Zahniser, M.S.; Kolb, C.E.; Ko, M.K.W.; Weisenstein, D.K. OH reaction kinetics and atmospheric impact of 1-bromopropane. J. Phys. Chem. 1997, 101, 4987–4990.
- Zetzsch, C.; Behnke, W. Heterogeneous photochemical sources of atomic Cl in the troposphere. Ber. Bunsenges. Phys. Chem. 1992, 96, 488–493. [Google Scholar] [CrossRef]
- Behnke, W.; Zetzsch, C. Formation of organohalogens from heterogeneous atmospheric chemistry simulated in an aerosol smog chamber. GDCh Monograph. 2005, 34, 110–121. [Google Scholar]
- Alligood, B.W.; FitzPatrick, B.L.; Szpunar, D.E.; Butler, L.J. Chloroacetone photodissoiation at 193 nm and the sub-sequent dynamics of the CHC(O)CH2 radical—An intermediate formed in the OH + allene reaction en route. J. Chem. Phys. 2011, 134, 054301. [Google Scholar] [CrossRef]
- Department of Water, Government of Western Australia. Waterway Assessment of the Upper Lockhart River: Camm River Confluence to Newdegate; Water resource management series, WRM 58; Avon Catchment Council: Northam, Australia, 2009.
- Department of Water, Government of Western Australia. Waterway Assessment of the Camm River: Lockhart River Confluence to Hyden; Water resource management series, WRM 57; Avon Catchment Council: Northam, Australia, 2009.
- Lillicrap, A.; George, R. The Distribution and Origins of Acid Groundwaters in the South West Agricultural Area; Resource Management Technical Report; Department of Agriculture and Food, Western Australia: Perth, Australia, 2010. [Google Scholar]
- Fitzpatrick, R.; Shand, P. Inland Acidic Sulfate Soil Systems across Australia; CRC LEME Open File Report; CRC LEME: Perth, Australia, 2008. [Google Scholar]
- Hookey, G.R. Prediction of delays in groundwater response to catchment clearing. J. Hydrol. 1987, 94, 181–198. [Google Scholar] [CrossRef]
- Nulsen, R.A. Catchment Hydrology Group. Groundwater Trends in the Agricultural Area of Western Australia; Resource Management Technical Report No. 173; Agriculture Western Australia: Perth, Australia, 1998. [Google Scholar]
- Addison, D. Groundwater Study of the Lake Grace Townsite; Resource Management Technical Report 212; Agriculture Western Australia: Perth, Australia, 2001. [Google Scholar]
- Ghauri, S. Groundwater Trends in the Central Agricultural Region; Resource Management Technical Report 269; Department of Agriculture, Western Australia: Perth, Australia, 2004. [Google Scholar]
- Ruprecht, J.K.; Schofield, N.J.S. Effects of partial deforestation on hydrology and salinity in high salt storage landsapes. II strip, soil and parkland clearing. J. Hydrol. 1991, 129, 39–55. [Google Scholar] [CrossRef]
- Rogers, S.; George, R. WA Wheatbelt drainage—Acidic groundwater, not just a salt issue. Focus Salt 2005, 33, 8–9. [Google Scholar]
- Degens, B.; Shand, P. Assessment of acidic saline groundwater hazard in the Western Australian Wheatbelt: Yarra Yarra, Blackwood and South Coast; Water for a Healthy Country National Research Flagship; CSRIO: Perth, Australia, 2010. [Google Scholar]
- Mann, A.W. Hydrogeochemistry and weathering on the Yilgarn Block, Western Australia—Ferrolysis and heavy metals in continental brines. Geochim. Cosmochim. Acta 1983, 47, 181–190. [Google Scholar] [CrossRef]
- McArthur, J.M.; Turner, J.V.; Lyons, W.B.; Osborn, A.O.; Thirlwall, M.F. Hydrochemistry on the Yilgarn Block, Western Australia: Ferrolysis and mineralization in acidic brines. Geochim. Cosmochim. Acta 1991, 55, 1273–1288. [Google Scholar] [CrossRef]
- Shand, P.; Degens, B. Avon Catchment Acidic Groundwater—Geochemical Risk Assessment; CRC LEME Open File Report; CRC LEME: Perth, Australia, 2008; p. 82. [Google Scholar]
- Kamilli, K.A.; Ofner, J.; Krause, T.; Sattler, T.; Schmitt-Kopplin, P.; Eitenberger, E.; Friedbacher, G.; Lendl, B.; Lohninger, H.; Schöler, H.F.; et al. How salt lakes affect atmospheric new particle formation: A case study in Western Australia. Sci. Total Environ. 2016, 573, 985–995. [Google Scholar] [CrossRef]
- Junkermann, W.; Hacker, J.; Lyons, T.; Nair, U. Land use change supresses precipitation. Atmos. Chem. Phys. 2009, 9, 6531–6539. [Google Scholar] [CrossRef]
- Kamilli, K.A.; Ofner, J.; Lendl, B.; Schmitt-Kopplin, P.; Held, A. New particle formation above a simulated salt lake in aerosol chamber experiment. Environ. Chem. 2015, 12, 504–514. [Google Scholar] [CrossRef]
- Cocker, D.R.; Flagan, R.C.; Seinfeld, J.H. State-of-the-art chamber facility for studying atmospheric aerosol chemistry. Environ. Sci. Technol. 2001, 35, 2594–2601. [Google Scholar] [CrossRef]
- Toyota, K.; Kanaya, Y.; Takahashi, M.; Akimoto, H. A box model study on photochemical interactions between VOCs and reactive halogen species in the marine boundary layer. Atmos. Chem. Phys. 2004, 4, 1961–1987. [Google Scholar] [CrossRef] [Green Version]
- Bleicher, S. Zur Halogenaktivierung im Aerosol und in Salzpfannen 2012. Ph.D. Thesis, Universität Bayreuth, Bayreuth, Germany, 2012. [Google Scholar]
- Bleicher, S.; Buxmann, J.; Sander, R.; Riedel, T.P.; Thornton, J.A.; Platt, U.; Zetzsch, C. The influence of nitrogen oxides on the activation of bromide and chloride in salt aerosol. Atmos. Chem. Phys. Discuss. 2014, 14, 10135–10166. [Google Scholar] [CrossRef]
- Buxmann, J.; Balzer, N.; Bleicher, S.; Platt, U.; Zetzsch, C. Observations of bromine explosions in smog chamber experiments above a model salt pan. Int. J. Chem. Kinet. 2012, 44, 312–326. [Google Scholar] [CrossRef]
- Orlando, J.J. Temperature dependence of the rate coeffients for the reaction of chlorine atoms with chloromethanes. Int. J. Chem. Kinet. 1999, 31, 515–524. [Google Scholar] [CrossRef]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.G.; Pilling, M.J. Protocol for the development of the master chemical mechanism, MCM v3 (part: A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef]
- NCAR Earth System Laboratory, Atmospheric Chemistry Division. Available online: http://cprm.acom.ucar.edu/Models/TUV/Interactive_TUV/ (accessed on 13 September 2019).
- Physick, W.L.; Tapper, N.J. A numerical study of circulations induced by a dry salt lake. Mon. Wea. Rev. 1990, 118, 1029–1042. [Google Scholar] [CrossRef]
- Keller-Rudek, H.; Moortgat, G.K.; Sander, R.; Sörensen, R. The MPI-Mainz UV/VIS spectral atlas of gaseous molecules of atmospheric interest. Earth Syst. Sci. Data 2013, 5, 365–373. [Google Scholar] [CrossRef]
- The MPI_Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest. Available online: www.uv-vis-spectral-atlas-mainz.org (accessed on 13 September 2019).
- Kopetzky, R. Bildung halogenierter organischer Verbindungen aus salzhaltigen Lösungen 2005. Diploma Thesis, Universität Lüneburg, Lüneburg, Germany, 2005. [Google Scholar]
- Sörgel, M. Experimentelle Untersuchung zur Bildung halogenorganischer Verbindungen aus Huminsäuren in Abhängigkeit vom pH-Wert 2007. Ph.D. Thesis, Hochschule für Angewandte Wissenschaften Hamburg, Hamburg, Germany, 2007. [Google Scholar]
- Yamada, E.; Doi, K.; Okano, K.; Fuso, Y. Simultaneous determination of the concentration and molecular weight of humic substances in environmental water by gel chromatography with fluorescence detector. Anal. Sci. 2000, 16, 125–129. [Google Scholar] [CrossRef]
- Levene, P.A. Bromoacetone. Org. Synth. 1930, 10. [Google Scholar] [CrossRef]
- Hodgeson, J.W.; Cohen, A.I. Determination of Chlorinated Desinfection Byproducts and Chlorinated Solvents in Drinking Water by Liquid-Liquid Extraction and Gas Chromatography with Electron-Capture Detection; Environmental Monitoring Systems Laboratory Office of Research and Development, U.S. Environmental Protection Agency: Cincinnati, OH, USA, 1990. [Google Scholar]
- Kolb, B. Application of gas chromatographic head-space analysis for the characterization of non-ideal solution by scanning the total concentration range. J. Chromatogr. A 1975, 112, 287–295. [Google Scholar] [CrossRef]
- Kolb, B.; Welter, C.; Bichler, C. Determination of partition coefficients by automatic equilibrium headspace gas chromatography by vapour phase calibration. Chromatographia 1992, 34, 235–240. [Google Scholar] [CrossRef]
- Kolb, B.; Ettre, L.S. Static Headspace-Gas Chromatography: Theory and Practice; Wiley-VCH: New York, NY, USA, 2006. [Google Scholar]
- Stutz, J.; Ackermann, R. Atmospheric reactive chlorine and bromine at the Greate Salt Lake. Utha. Geophys. Res. Lett. 2002, 29, 197–256. [Google Scholar] [CrossRef]
- Wingenter, O.W.; Sive, B.C.; Blake, N.J.; Blake, D.R.; Rowland, F.S. Atomic chlorine concentrations derived from ethane and hydroxyl measurements over the equatorial Pacific Ocean: Implication for dimethyl sulfide and bromine monoxide. J. Geophys. Res. 2005, 110, D20308. [Google Scholar] [CrossRef]
- Wittmer, J.; Bleicher, S.; Zetzsch, C. Iron(III)-induced activation of chloride and bromide from modeled salt pans. J. Phys. Chem. A 2014, 119, 4373–4385. [Google Scholar] [CrossRef]
- Palm, W.-U.; Millet, M.; Zetzsch, C. Photochemicals reaction of metamitron. Chemosphere 1997, 35, 117–1130. [Google Scholar] [CrossRef]
- Meylan, W.M.; Howard, P.H. Bond contribution method for estimating Henry’s law constants. Environ. Toxicol. Chem. 1991, 10, 1283–1293. [Google Scholar] [CrossRef]
- Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Betterton, E.A. The partitioning of ketones between the gas and aqueous phases. Atmos. Environ. Part A. Gen. Top. 1991, 25.8, 1473–1477. [Google Scholar] [CrossRef]
- Peters, R.J.B.; de Leer, E.W.B.; Versteegh, J.F.M. Identification of halogenated compounds produced by chlorination of humic acid in the presence of bromide. J. Chromatogr. 1994, 686, 253–261. [Google Scholar] [CrossRef]
- Kieber, R.J.; Zhou, X.; Mopper, K. Formation of carbonyl compounds from UV-induced photodegradation of humic substances in natural waters: Fate of riverine carbon in the sea. Limnol. Oceanogr. 1990, 35, 1503–1515. [Google Scholar] [CrossRef]
- Zhou, X.; Mopper, K. Photochemical production of low-molecular-weight carbonyl compounds in seawater and surface microlayer and their air-sea exchange. Marine Chem. 1997, 56, 201–213. [Google Scholar] [CrossRef]
- Westerhoff, P.; Chao, P.; Mash, H. Reactivity of natural organic matter with aqueous chlorine and bromine. Water Res. 2004, 38, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Deborde, M.; von Gunten, U. Reactions of chlorine with inorganic and organic compounds during water treatment-Kinetics and mechanisms: A critical review. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef] [PubMed]
- HSDB (2015): Hazardous Substances Data Bank (HSDB)–Toxnet–National Institute of Health, National Library of Medicine, 8600 Rockville Pike, Bethesda, MD 20894 National Institutes of Health, Health & Human Services. Available online: https://toxnet.nlm.nih.gov/newtoxnet/hsdb.htm (accessed on 12 June 2015).
- Lewis, R.J. Hawley’s Condensed Chemical Dictionary, 15th ed.; Wiley: New York, NY, USA, 2007. [Google Scholar]
- Haag, W.R.; Hoigné, J. Ozonation of bromide-containing waters: Kinetics of formation of hypobromous acid and bromate. Environ. Sci. Technol. 1983, 17, 261–267. [Google Scholar] [CrossRef]
- Wittmer, J.; Zetzsch, C. Photochemical activation of chlorine by iron-oxide aerosol. J. Atmos. Chem. 2016, 73, 1–18. [Google Scholar] [CrossRef]
- Wittmer, J.; Bleicher, S.; Ofner, J.; Zetzsch, C. Iron(III)-induced activation of chloride from artificial sea salt aerosol. Environ. Chem. 2015, 12, 461–475. [Google Scholar] [CrossRef]
- Ofner, J.; Balzer, N.; Buxmann, J.; Grothe, H.; Schmitt-Kopplin, P.; Platt, U.; Zetzsch, C. Halogenation processes of secondary organic aerosol and implications on halogen release mechanisms. Atmos. Chem. Phys. 2012, 12, 5787–5806. [Google Scholar] [CrossRef] [Green Version]
- Behnke, W.; Elend, M.; Krüger, U.; Zetzsch, C. The influence of NaBr/NaCl ratio on the Br--catalysed production of halogenated radicals. J. Atmos. Chem. 1999, 34, 87–99. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Latitude (°) S | Longitude (°) E | Diameter (km) | pH | Amount of Air Samples |
|---|---|---|---|---|---|
| Lake Boats | −33.068538 | 119.636983 | 0.3 | 2.3 | 12 |
| Lake Bean | −33.161294 | 119.744744 | - | 7.1 | 8 |
| Lake Kathleen | −32.984241 | 119.694444 | - | 7 | 8 |
| Lake Orr | −33.148051 | 119.161199 | 0.6 | 4 | 4 |
| Lake Shot | −33.047973 | 119.610159 | - | 3.5 | 13 |
| Lake Strawbridge | −32.848018 | 119.396998 | 0.6 | 7.3 | 4 |
| Lake Dune | −33.084896 | 119.637903 | 0.3 | 2.9 | 15 |
| T (°C) | Bromoacetone + Cl− k ±σ (10−4 M−1s−1) | 1,1-Dibromoacetone + Cl− k ± σ (10−6M−1s−1) | Hydrolysis of Bromoacetone (10−6 M−1s−1) | Hydrolysis of 1,1-Dibromoacetone (10−6 M−1s−1) |
|---|---|---|---|---|
| 43.5 | 3.26 ± 0.13 | 4.2 ± 1.4 | ||
| 52.5 | 7.27 ± 0.09 | 14.0 ± 3.4 | ||
| 63.5 | 18.0 ± 0.55 | 73.5 ± 8.2 | ||
| 80.0 | - | - | 2.05 ± 0.33 | |
| 80.0 | 0.48 ± 0.21 |
| Compound | H313 | H323 | H333 | H343 | H353 | H363 | H373 |
|---|---|---|---|---|---|---|---|
| Bromoacetone | 47.4 ± 7.6 | 19.9 ± 5.7 | 15.3 ± 3.0 | 7.7 ± 1.5 | 7.0 ± 0.5 | 3.4 ± 0.5 | 2.7 ± 0.4 |
| 1,1-Dibromoacetone | 17.1 ± 2.5 | 9.09 ± 1.15 | 5.63 ± 0.31 | 3.20 ± 0.32 | 2.38 ± 0.15 | 1.38 ± 0.15 | 1.11 ± 0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sattler, T.; Sörgel, M.; Wittmer, J.; Bourtsoukidis, E.; Krause, T.; Atlas, E.; Benk, S.; Bleicher, S.; Kamilli, K.; Ofner, J.; et al. Natural Formation of Chloro- and Bromoacetone in Salt Lakes of Western Australia. Atmosphere 2019, 10, 663. https://doi.org/10.3390/atmos10110663
Sattler T, Sörgel M, Wittmer J, Bourtsoukidis E, Krause T, Atlas E, Benk S, Bleicher S, Kamilli K, Ofner J, et al. Natural Formation of Chloro- and Bromoacetone in Salt Lakes of Western Australia. Atmosphere. 2019; 10(11):663. https://doi.org/10.3390/atmos10110663
Chicago/Turabian StyleSattler, Tobias, Matthias Sörgel, Julian Wittmer, Efstratios Bourtsoukidis, Torsten Krause, Elliot Atlas, Simon Benk, Sergej Bleicher, Katharina Kamilli, Johannes Ofner, and et al. 2019. "Natural Formation of Chloro- and Bromoacetone in Salt Lakes of Western Australia" Atmosphere 10, no. 11: 663. https://doi.org/10.3390/atmos10110663
APA StyleSattler, T., Sörgel, M., Wittmer, J., Bourtsoukidis, E., Krause, T., Atlas, E., Benk, S., Bleicher, S., Kamilli, K., Ofner, J., Kopetzky, R., Held, A., Palm, W.-U., Williams, J., Zetzsch, C., & Schöler, H.-F. (2019). Natural Formation of Chloro- and Bromoacetone in Salt Lakes of Western Australia. Atmosphere, 10(11), 663. https://doi.org/10.3390/atmos10110663