Atmospheric Chemistry in a Box or a Bag

Abstract
1. Introduction
2. Requirements for Chamber Technology
2.1. Ideal Design Considerations
- Initial and endpoint concentrations of reactive species should approach those observed in the atmosphere.
- Initial input of gas mixtures (or aerosols) should be well-defined so that spurious reactions caused by reactant contamination are controlled entering the vessel.
- The light source for irradiation should simulate sunlight, or at a minimum, the light wavelengths known to be key for excitation of airborne species.
- The duration of an experiment should approach at least most of a day or a span of several hours.
- Air in the chambers should be well mixed to minimize concentration gradients in the chamber.
- Documentation and control of environmental variables like pressure, humidity and temperature should be assured or variability at least documented.
- Loss of reactants, degassing or byproducts of surface reactions at the vessel walls should be accounted for as an influence on the measured reactant-product profiles.
- Instrumentation for characterizing reactions should be as complete as practical from speciated VOCs and NOy (reactive nitrogen species) to oxidants, and particle physicochemical characteristics or other stable products and inference of free radical or other intermediate species.
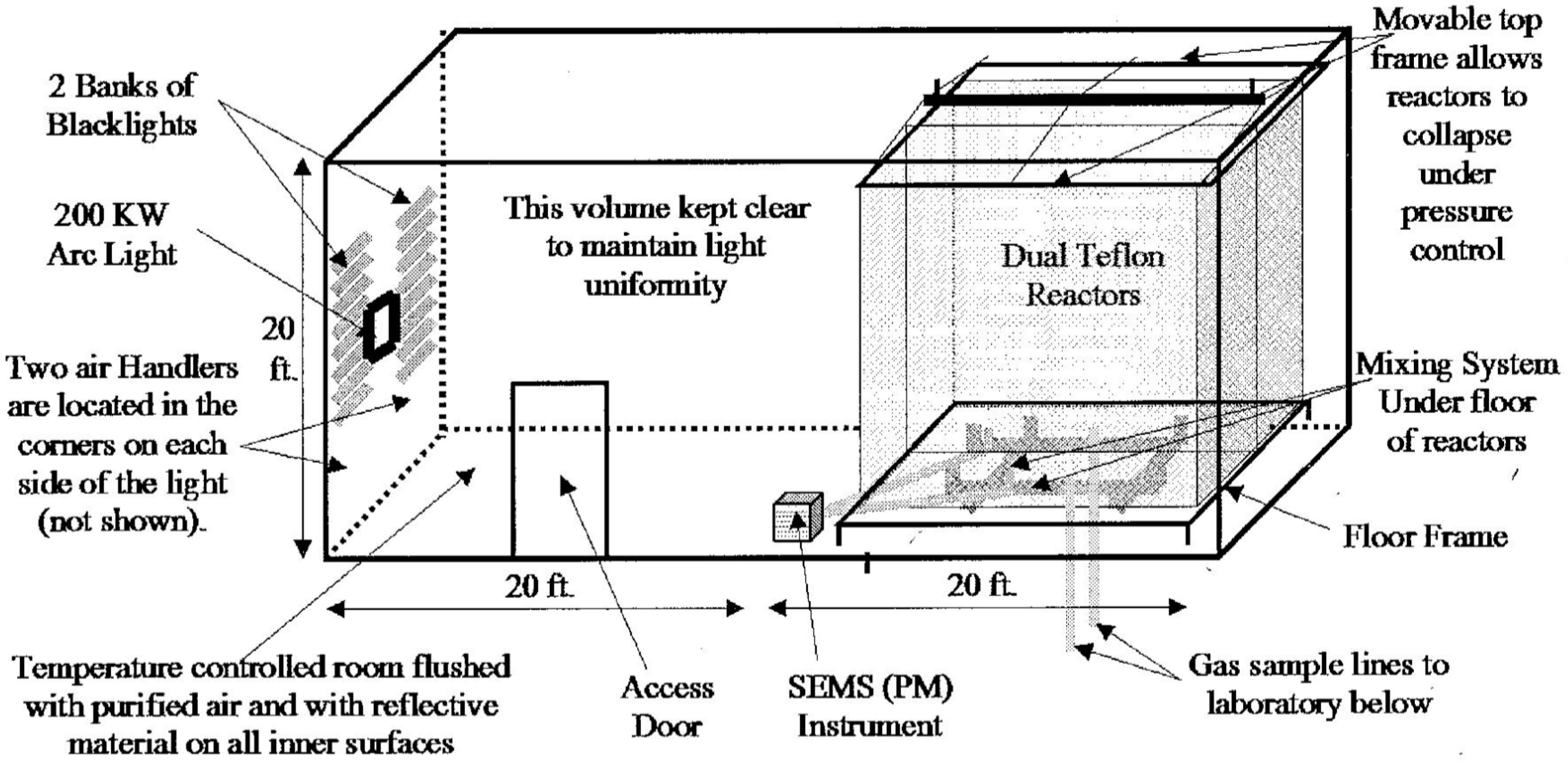
2.2. Some Chamber Facilities
2.3. Limitations of Laboratory Chemical Simulation
3. Knowledge Gained from Chambers
3.1. Atmospheric Photochemical Processes
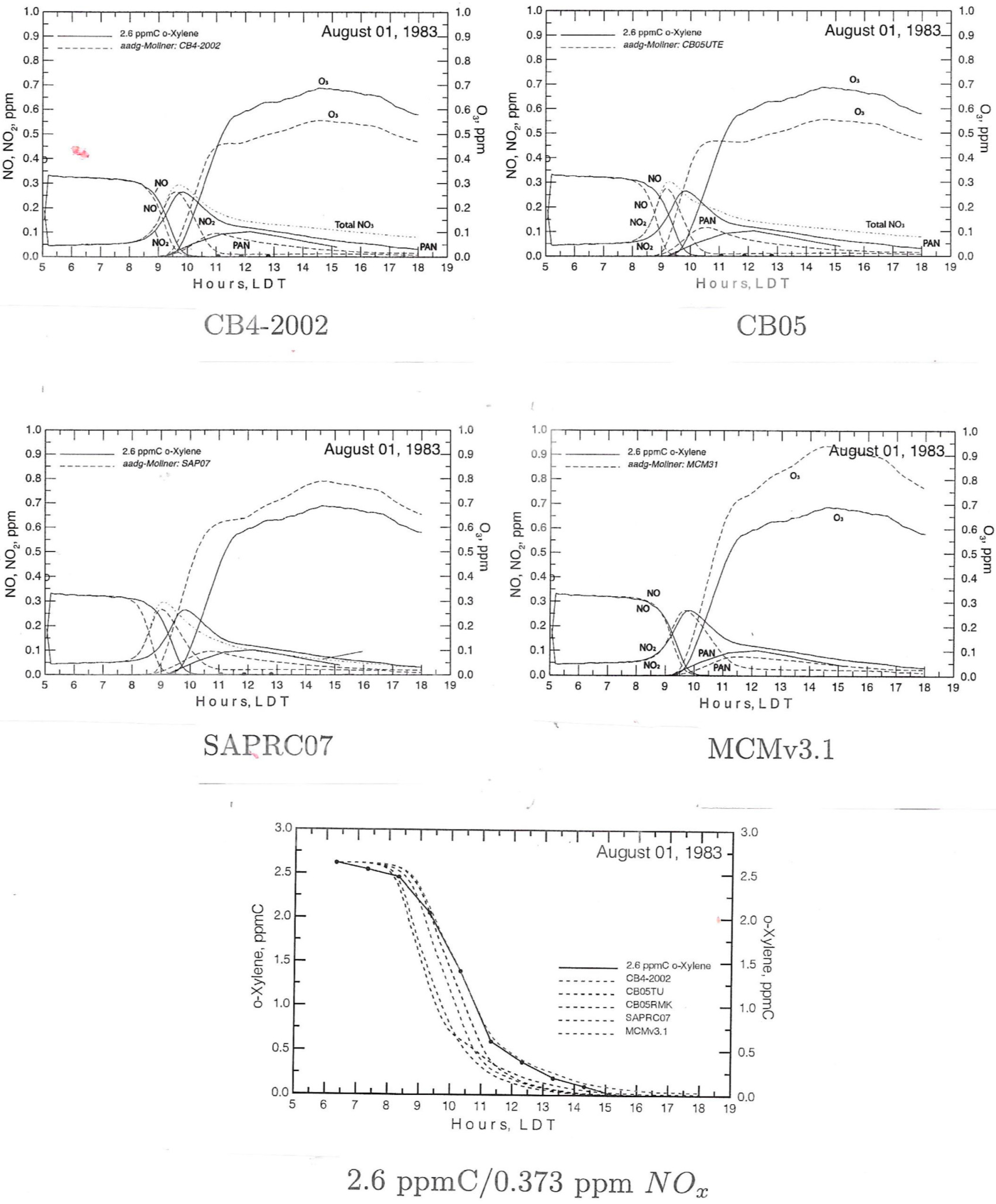
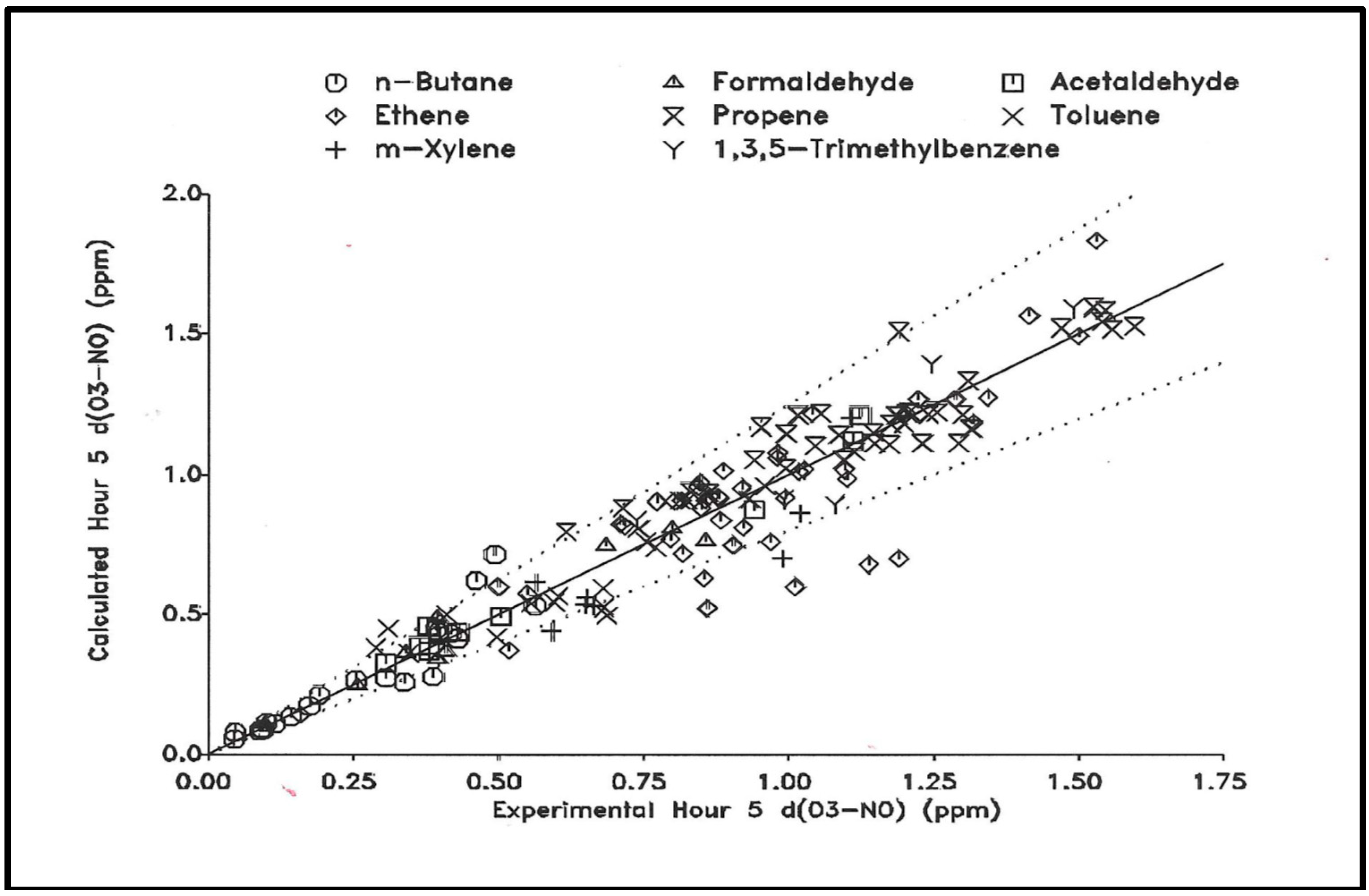
3.1.1. Mechanism evaluation
3.1.2. Relative Reactivity
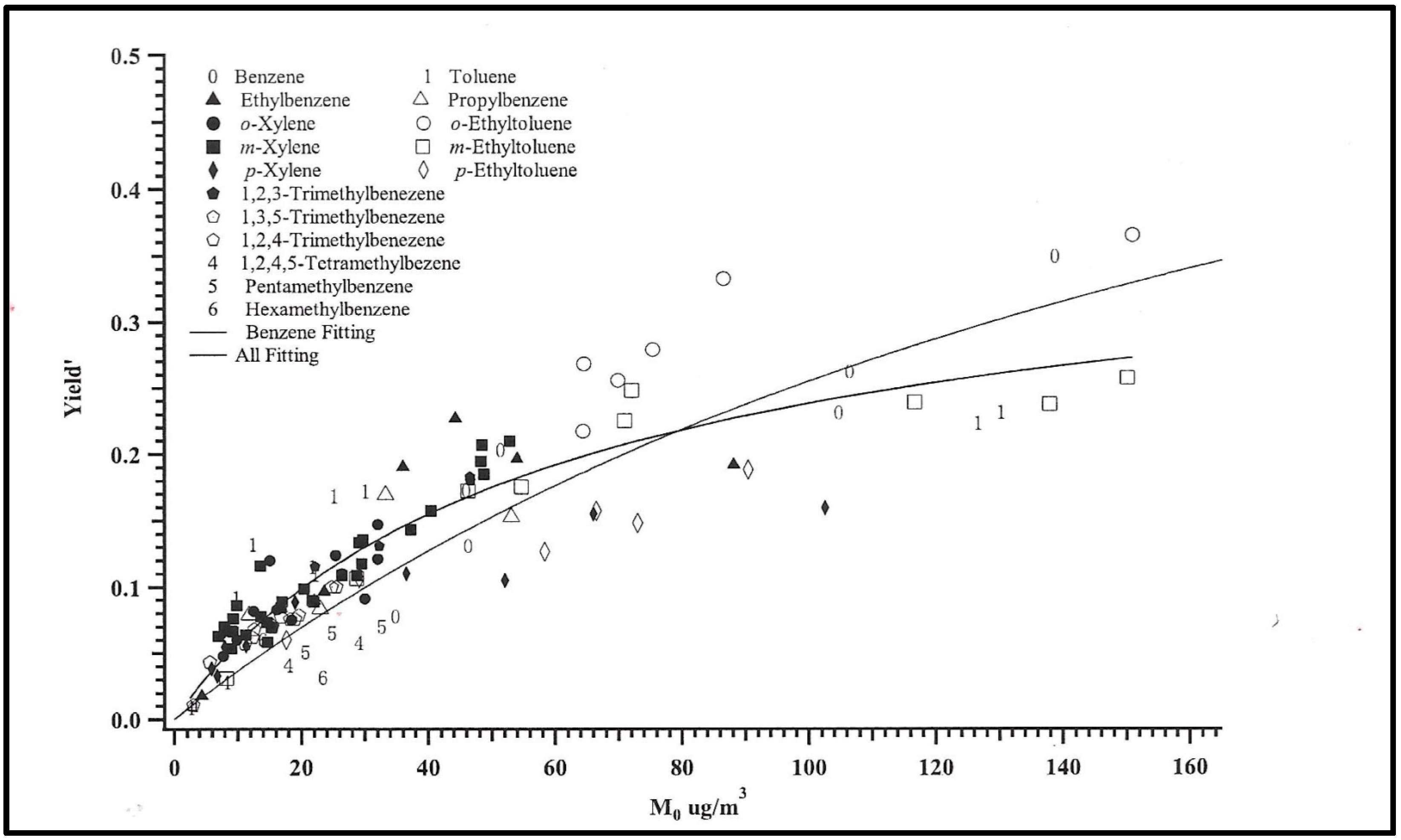
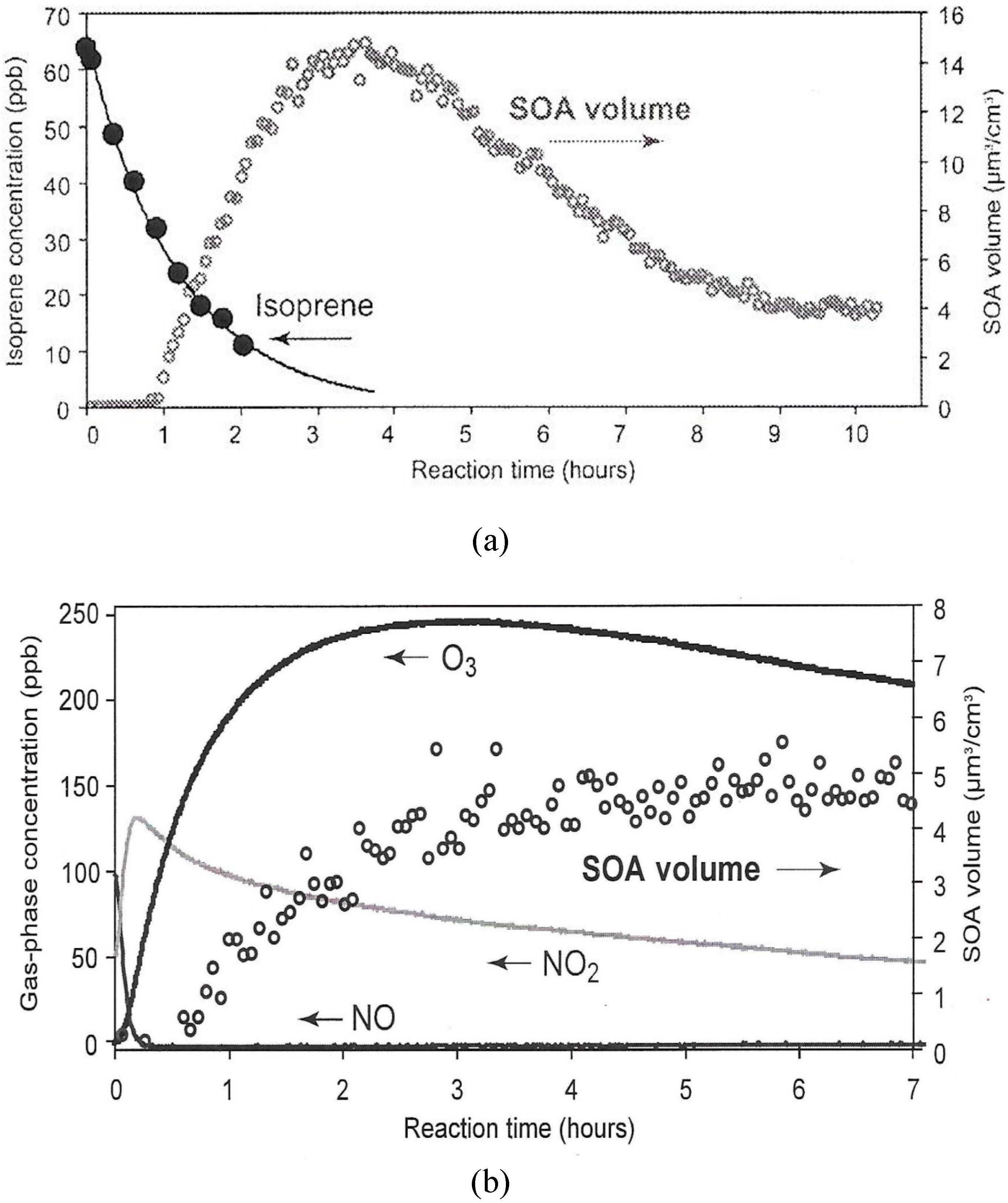
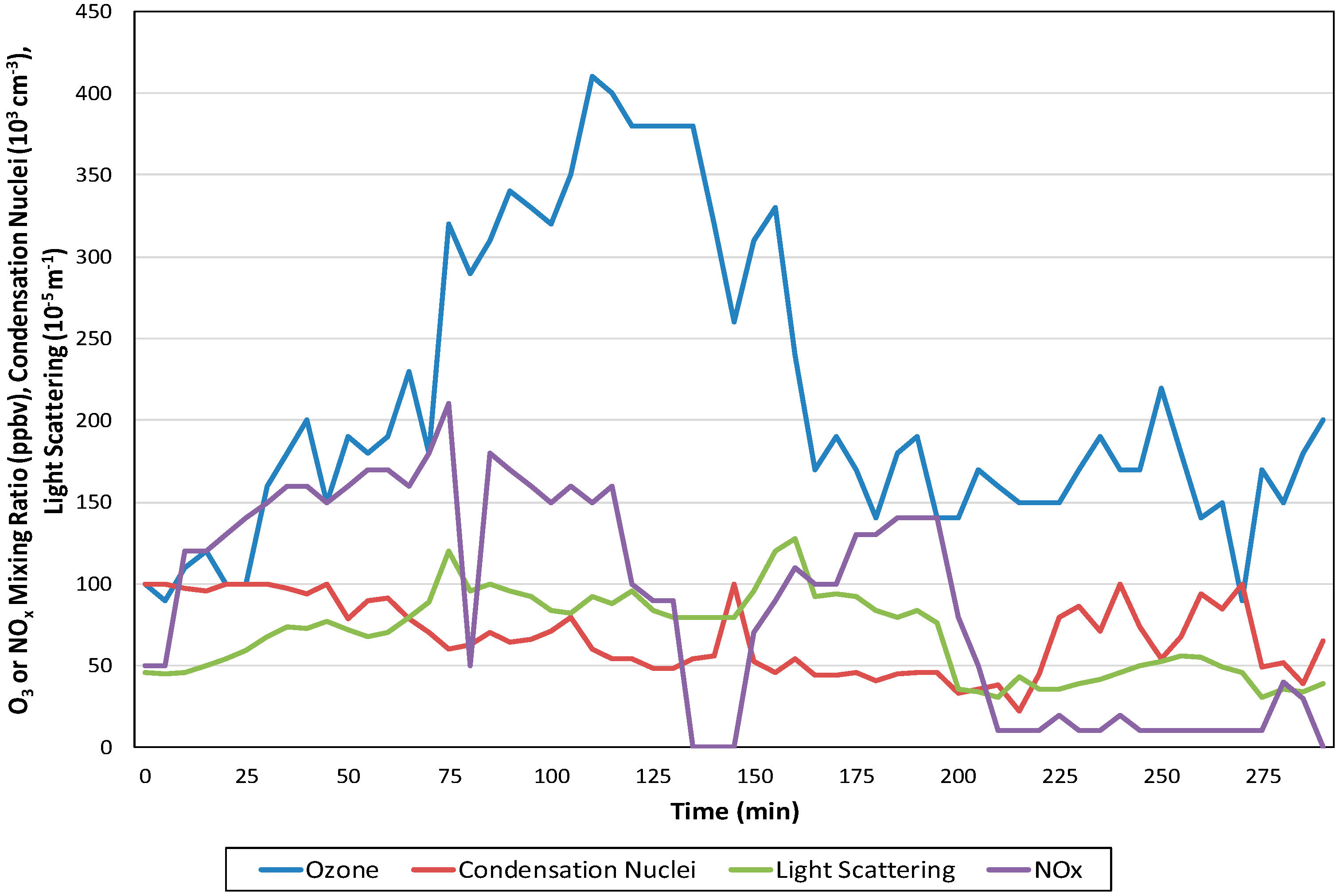
3.2. Photochemical Aerosols
SOA Thermo-kinetic Models
4. The Atmosphere—Judging the Simulation Quality
5. Summary
- --
- Focus on multiday gas and particle chemistry, including temperature and humidity interactions.
- --
- Conduct chamber and other experiments at concentrations approaching current levels atmospheric levels in rural and remote areas.
- --
- Strengthen aerosol models with kinetic mechanisms for condensed products.
- --
- Identify in more detail intermediate products including oxygenated species of gas-phase and particle reactions and identify added atmospheric tracer species for confirming laboratory mechanisms.
- --
- Examine and evaluate the potential importance of background conditions in the atmosphere as they may interact with the controlled experiments.
- --
- Give more attention to photochemistry of the free troposphere as a function of altitude, including hydrometeor interactions.
Funding
Acknowledgments
Conflicts of Interest
References
- Hidy, G.M. Adventures in Atmospheric Simulation. Bull. Am. Meteorol. Soc. 1967, 48, 143–161. [Google Scholar] [CrossRef][Green Version]
- Tyndall, J. On a new series of chemical reaction products by light. Proc. R. Soc. Lond. 1868, 17, 92–102. [Google Scholar]
- Wilson, C.T.R.; Grimes, R.W.; Catlow, C.R.A. Condensation of Water Vapour in the Presence of Dust-Free Air and Other Gases. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1897, 189, 265–307. [Google Scholar] [CrossRef]
- Magill, P. Techniques Employed in the Analysis of Los Angeles Smog. In Proceedings of the First National Air Pollution Symposium, Los Angeles, CA, USA, 10–11 November 1949; pp. 61–68. [Google Scholar]
- Haagen-Smit, A.; Fox, M. Ozone formation in Photochemical Oxidation of Organic Substances. Ind. Eng. Chem. 1956, 48, 1484–1487. [Google Scholar] [CrossRef]
- Renzetti, N. Ozone in Los Angeles Atmosphere. In Ozone Chemistry and Technology. Adv. Chem. 1959, 21, 230–282. [Google Scholar]
- Kehoe, R. Air Pollution and Community Health. In Proceedings of the First National Air Pollution Symposium, Los Angeles, CA, USA, 10–11 November 1949; pp. 115–120. [Google Scholar]
- Renzetti, N.; Doyle, G. The chemical nature of particulate in irradiated automobile exhaust. J. Air Pollut. Control Assoc. 1959, 8, 293–296. [Google Scholar] [CrossRef][Green Version]
- Stephens, E.; Schuck, E. Air pollution effects of irradiated auto exhaust as related to fuel consumption. Chem. Eng. Prog. 1958, 54, 71–77. [Google Scholar]
- Schuck, E.; Doyle, G. Photo-Oxidation of Hydrocarbons in Mixtures Containing Oxides of Nitrogen and Sulfur Dioxide; Report 29; Air Pollution Foundation: Los Angeles, CA, USA, 1959. [Google Scholar]
- Mader, P.; MacPhee, R.; Lafberg, R.; Larson, C. Composition of the organic part of atmospheric aerosols in Los Angeles. Ind. Eng. Chem. 1952, 44, 1352–1355. [Google Scholar] [CrossRef]
- Mailila, J. On the early studies recognizing the role of sulfuric acid atmospheric haze and new particle formation. Tellus 2018, 70, 1–11. [Google Scholar] [CrossRef]
- Richards, L. The Formation of Ozone in Polluted Atmospheres. J. Air Pollut. Control Assoc. 1956, 5, 216–246. [Google Scholar] [CrossRef]
- Hamming, W.; Dickinson, J. Control of Photochemical Smog by Alteration of Initial Reactant Ratios. J. Air Pollut. Control Assoc. 1966, 16, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Romanovksy, J.C.; Ingels, R.; Gordon, R. Estimation of Smog Effects in the Hydrocarbon-Nitric Oxide System. J. Air Pollut. Control Assoc. 1967, 17, 454–459. [Google Scholar] [CrossRef]
- Dimitriades, B. EPA Report 600/3-76-029; US Environmental Protection Agency: Research Triangle Park, NC, USA, 2005.
- Dimitriades, B. Methodology in Air Pollution Studies Using Irradiation Chambers. J. Air Pollut. Control Assoc. 1967, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Harkins, J.; Nicksic, S. Studies of the Role of Sulfur Dioxide in Visibility Reduction. J. Air Poll. 1965, 15, 218–221. [Google Scholar] [CrossRef]
- Stephens, E.; Price, M. Comparison of Synthetic and Smog Aerosols. J. Colloid Interface Sci. 1972, 39, 272–286. [Google Scholar] [CrossRef]
- Wilson, W.; Merryman, E.; Levy, A.; Taliaferro, H. Aerosol Formation in Photochemical Smog. J. Air Pollut. Control Assoc. 1971, 21. [Google Scholar] [CrossRef]
- Wilson, W.; Levy, A.; Wimmer, D.A. Study of Sulfur Dioxide in Photochemical Smog. J. Air Pollut. Control Assoc. 1972, 22, 27–32. [Google Scholar] [CrossRef]
- Kocmond, W.; Yang, J.; Kittleson, S.; Whitby, K.; Demerjian, K. Aerosol Formation in Simple Photochemical Systems. In Fate of Pollutants in the Air and Water Environments; Part 2: Chemical and Biological Fate of Pollutants in Environment; Suffet, I., Ed.; Wiley-Interscience: New York, NY, USA, 1977; pp. 101–134. [Google Scholar]
- O’Brien, R.; Holmes, J.; Bockian, A. Formation of Photochemical Aerosols from Hydrocarbons. Environ. Sci. Technol. 1975, 9, 568–576. [Google Scholar] [CrossRef]
- Heisler, S.; Friedlander, S. Gas-to-particle conversion in photochemical smog: Aerosol growth laws and mechanisms for organics. Atmos. Environ. 1977, 11, 157–168. [Google Scholar] [CrossRef]
- Grosjean, D.; Friedlander, S. Formation of Organic Aerosols from Cyclic Olefins and Diolefins. In The Character and Origins of Smog Aerosols; Hidy, G., Mueller, P., Grosjean, D., Appel, B., Wesolowski, J., Eds.; Wiley-Interscience: New York, NY, USA, 1980; pp. 435–476. [Google Scholar]
- Leighton, P. Photo Chemistry of Air Pollution; Academic Press: New York, NY, USA, 1961. [Google Scholar]
- A Theory of Upper-Atmospheric Ozone. Available online: https://www.rmets.org/sites/default/files/papers/chapman-memoirs.pdf (accessed on 10 July 2019).
- Finlayson-Pitts, B.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere; Academic Press: New York, NY, USA, 2000. [Google Scholar]
- Seinfeld, J.; Pandis, S. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; Wiley-Interscience: New York, NY, USA, 2006. [Google Scholar]
- Paul Scherrer Institute. Smog Chamber Description. Available online: www.psich/lac/smog-chamber (accessed on 4 April 2019).
- Bae, G.; Kim, M.; Lee, S.; Song, K.; Jin, H.; Moon, K. Design and performance evaluation of the KIST indoor smog chamber. J. Korean Soc. Atmos. Environ. 2003, 19, 437–449. [Google Scholar]
- Becker, K. Overview on the Development of Chambers for the Study of Atmospheric Chemical Processes. In Environmental Simulation Chambers: Application to Atmospheric Chemical Processes; Barnes, I., Rudzinski, K., Eds.; Springer: New York, NY, USA, 2006; pp. 1–26. [Google Scholar]
- Wagner, R.; Bunz, H.; Linke, C.; Mohler, O.; Naumann, K.-H.; Saathoff, H.; Schnaiter, M.; Schurath, U. Chamber Simulations of Cloud Chemistry: The AIDA Chamber. In Environmental Simulation Chambers: Application to Atmospheric Chemical Processes; Barnes, I., Rudzinski, K., Eds.; Springer: New York, NY, USA, 2006; pp. 67–82. [Google Scholar]
- Wang, X.; Liu, T.; Bernard, F.; Ding, X.; Wen, S.; Zhang, Y.; He, Q.; Lu, S.; Chen, J.; Saunders, S.; et al. Design and characterizations of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation. Atmos. Meas. Tech. 2014, 7, 301–313. [Google Scholar] [CrossRef]
- Babar, Z.; Park, J.-H.; Kang, J.; Lim, H.J. Characterization of a Smog Chamber for Studying Formation and Physicochemical Properties of Secondary Organic Aerosol. Aerosol Air Qual. Res. 2016, 16, 3102–3113. [Google Scholar] [CrossRef]
- Dodge, M. Chemical oxidant mechanisms for air quality modeling: Critical review. Atmos. Environ. 2000, 34, 2103–2130. [Google Scholar] [CrossRef]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. Discuss. 2009, 9, 3555–3762. [Google Scholar] [CrossRef]
- Atkinson, R. Atmospheric Chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. [Google Scholar] [CrossRef]
- George, C.; Ammann, M.; D’Anna, B.; Donaldson, D.; Nizkorodov, S. Heterogeneous Photochemistry in the Atmosphere. Chem. Rev. 2015, 115, 318–4258. [Google Scholar] [CrossRef]
- Kroll, J.; Seinfeld, J. Chemistry of Secondary Organic Aerosol Formation and Evolution of Low Volatility Organics in the Atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Carlton, A.; Wiedinmyer, C.; Kroll, J. A review of secondary organic aerosol (SOA) formation from isoprene. Atmos. Chem. Phys. 2009, 9, 4987–5005. [Google Scholar] [CrossRef]
- Lim, Y.; Tan, Y.; Perri, M.; Serizinger, S.; Turpin, B. Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Atmos. Chem. Phys. 2010, 10, 10521–10539. [Google Scholar] [CrossRef]
- Stockwell, W.; Lawson, C.; Saunders, E.; Goliff, W. A Review of Tropospheric Chemistry and Gas-Phase Mechanisms for Air Quality Modeling. Atmosphere 2012, 3, 1–32. [Google Scholar] [CrossRef]
- Glasius, M.; Goldstein, A. Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry. Environ. Sci. Technol. 2016, 50, 2654–2764. [Google Scholar] [CrossRef]
- Bianchi, F.; Kurten, T.; Riva, M.; Mohr, C.; Rissanen, M.; Roldin, P.; Berndt, T.; Crounse, J.; Wennberg, P. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Chem. Rev. 2019, 119, 3472–3509. [Google Scholar] [CrossRef] [PubMed]
- Lipeles, M.; Landis, D.; Hidy, G. The Formation of Organic Aerosols in a Fast Flow Reactor. In Fate of Pollutants in the Air and Water Environment, Part II; Suffet, I., Ed.; John Wiley and Sons: New York, NY, USA, 1977; pp. 69–94. [Google Scholar]
- Franzblau, E.; Burton, C.; Hidy, G. Aerosol Particle Formation from Ozone-Terminal Olefin Reactions. Aerosol Sci. Technol. 1984, 3, 167–176. [Google Scholar] [CrossRef]
- Lambe, A.; Ahern, A.; Williams, L.; Slowik, J.; Wong, J.; Abbatt, J.; Brune, W.; Ng, N.; Wright, J.; Croasdale, D.; et al. Characterization of aerosol photooxidation reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Atmos. Meas. Tech. 2011, 4, 445–461. [Google Scholar] [CrossRef]
- Kang, E.; Root, M.; Toohey, D.; Brune, W. Introducing the concept of Potential Aerosol Mass (PAM). Atmos. Chem. Phys. 2007, 7, 5727–5744. [Google Scholar] [CrossRef]
- Sickles, J., II; Ripperton, L.; Eaton, W.; Wright, R. Oxidant-Precursor Relationships under Pollutant Transport Conditions; EPA Report 600/3-79-078a; US Environmental Protection Agency: Research Triangle Park, NC, USA, 1979.
- Jeffries, H.; Fox, D.; Kamens, D. Outdoor smog chamber studies: Light effects relative to indoor chambers. Environ. Sci. Technol. 1976, 10, 1006–1011. [Google Scholar] [CrossRef]
- White, S.; Angove, D.; Li, K.; Campbell, I.; Element, A.; Halliburton, B.; Lavrencic, S.; Cameron, D.; James, I.; Azzi, M. Development of a new smog chamber for studying the impact of different uv lamps on SAPRC chemical mechanisms predictions and aerosol formation. Environ. Chem. 2018, 15, 171–182. [Google Scholar] [CrossRef]
- Vera, T.; Munoz, A.; Rodenas, M.; Vazquez, M.; Mellouki, A.; Treacy, J.; Al Mulla, I.; Sidebottom, H. Photolysis of Trichloronitromethane (Chloropirin) under Atmospheric Conditions. Z. Phys. Chem. 2010, 224, 1039–1057. [Google Scholar] [CrossRef]
- Darnall, K.; Atkinson, R.; Winer, A.; Pitts, J., Jr. Effects of Constant vs. Diurnally-Varying Light Intensity on Ozone Formation. J. Air Pollut. Control Assoc. 1981, 31, 262–264. [Google Scholar] [CrossRef][Green Version]
- Dodge, M.; Bufalini, J.; Kopczynski, S. Contamination in Smog Chambers. Environ. Lett. 1972, 3, 101–109. [Google Scholar]
- Bufalini, J.; Theodore, A.; Marijon, M. Contamination Effects in O3 Formation in Smog Chambers. Environ. Sci. Technol. 1977, 11, 1181–1185. [Google Scholar] [CrossRef]
- Lonneman, W.; Bufalini, J.; Kuntz, T.; Meeks, S. Contamination from Fluorocarbon Films. Environ. Sci. Technol. 1981, 15, 99–103. [Google Scholar] [CrossRef]
- Kelly, N.; Olson, K.; Wong, C. Tests for Fluorocarbon and other Organic Vapor Release by Fluorocarbon Bags. Environ. Sci. Technol. 1985, 19, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, D. Wall loss of Gaseous Pollutants in Outdoor Teflon Chambers. Environ. Sci. Technol. 1985, 19, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, G.; Heo, G.; Carter, W.; Whitten, G. Environmental Chamber Experiments to Evaluate NOx Sinks and Recycling in Atmospheric Chemical Mechanisms; Report 06-2569981; Environ: Novaro, CA, USA, 2012. [Google Scholar]
- Trump, E.; Epstein, S.; Riipinen, I.; Donohue, N. Wall effects in smog chamber experiments: A model study. Aerosol Sci. Technol. 2016, 50, 1180–1200. [Google Scholar] [CrossRef]
- Akimoto, H.; Hoshimo, M.; Inoue, G.; Sakamaki, F.; Washida, N.; Okuda, M. Design and Characterization of the Evacuable and Bakable Photochemical Smog Chamber. Environ. Sci. Technol. 1979, 13, 471–475. [Google Scholar] [CrossRef]
- Carter, W.; Atkinson, E.; Winer, A.; Pitts, J., Jr. Evidence for Chamber-Dependent Radical Sources: Impact on Kinetic Computer Models for Air Pollution. Int. J. Chem. Kinet. 1981, 13, 735–740. [Google Scholar] [CrossRef]
- Carter, W.; Atkinson, R.; Winer, A.; Pitts, J., Jr. Experimental investigation of chamber-dependent radical sources. Int. J. Chem. Kinet. 1982, 14, 1071–1103. [Google Scholar] [CrossRef]
- Carter, W.; Atkinson, R.; Winer, A.; Pitts, J., Jr. The Wall as a Source of Hydroxl Radicals in Smog Chambers. Atmos. Environ. 1985, 19, 1977–1978. [Google Scholar] [CrossRef]
- Besemer, A.; Nieboer, H. The wall as a source of hydroxyl radicals in smog chambers. Atmos. Environ. 1985, 19, 507–513. [Google Scholar] [CrossRef]
- Sasamaki, F.; Akimoto, H. HONO Formation as Unknown Radical Source in Photochemical Smog Chamber. Int. J. Chem. Kinet. 1988, 20, 111–116. [Google Scholar] [CrossRef]
- Rohrer, F.; Bohn, B.; Brauers, T.; Bruning, D.; Johnen, F.-J.; Wahner, A.; Kleffmann, J. Characterisation of the photolytic HONO-source in the atmospheric simulation chamber SAPHIR. Atmos. Chem. Phys. 2005, 5, 2189–2201. [Google Scholar] [CrossRef]
- Iraci, L.; Johnston, J.; Golden, D. Experimental Techniques for Studying Surface Chemistry in Smog Chambers. Available online: www. cert.ucr.edu/-carter/epacham/iraci.pdf (accessed on 10 April 2019).
- McMurry, P.; Rader, D. Aerosol losses in electrically charged chambers. Aerosol Sci. Technol. 1985, 4, 249–268. [Google Scholar] [CrossRef]
- Charan, S.; Kong, W.; Flagan, R.; Seinfeld, J. Effect of particle charge on aerosol dynamics in Teflon environmental chambers. Aerosol Sci. Technol. 2018, 52, 854–871. [Google Scholar] [CrossRef]
- McMurry, P.; Grosjean, D. Gas and Aerosol Wall Losses in Teflon Film Smog Chambers. Environ. Sci. Technol. 1985, 19, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Ding, X.; Hakala, J.; Hofbauer, V.; Robinson, E.; Donahue, N. Vapor wall loss of semi-volatile organic compounds in a Teflon chamber. Aerosol Sci. Technol. 2016, 50, 822–9343. [Google Scholar] [CrossRef]
- Matsunaga, A.; Ziemann, P. Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci. Technol. 2010, 44, 881–892. [Google Scholar] [CrossRef]
- Krechmer, J.; Pagonis, D.; Ziemann, P.; Jiminez, J. Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low Volatility Oxidized Species. Environ. Sci. Technol. 2016, 50, 5757–5765. [Google Scholar] [CrossRef]
- Zhang, X.; Cappa, C.; Jathar, S.; McVay, R.; Ensberg, J.; Kleeman, M.; Seinfeld, J. Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol. Proc. Natl. Acad. Sci. USA 2014, 111, 5802–5807. [Google Scholar] [CrossRef]
- Bian, Q.; May, A.; Kreidenweis, S.; Pierce, J. Investigation of particle and vapor wall-loss effects on controlled wood-smoke smog-chamber experiments. Atmos. Chem. Phys. 2015, 15, 11027–11045. [Google Scholar] [CrossRef]
- Seinfeld, J. Effect of Vapor Loss in Laboratory Chambers on Yields of Secondary Organic Aerosols; Final Report to the California Air Resources Board; California Air Resources Board: Sacramento, CA, USA, 2015.
- Krechmer, J.; Day, D.; Ziemann, P.; Jiminez, J. Direct Measurements of Gas/Particle Partitioning and Mass Accomodation Coefficients in Environmental Chambers. Environ. Sci. Technol. 2017, 51, 11867–11875. [Google Scholar] [CrossRef] [PubMed]
- Brune, W. The Chamber Wall Index for Gas-Wall Interactions in Atmospheric Environmental Enclosures. Environ. Sci. Technol. 2019, 53, 3645–3652. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.; Englehart, G.; Hildebrandt, L.; Weitkamp, E.; Parthak, E.; Donahue, N. Constraining Particle Evolution from Wall Losses, Coagulation and Condensation-Evaporation in Smog Chamber Experiments: Optimal Based Size Distribution Measurements. Aerosol Sci. Technol. 2008, 42, 1001–1015. [Google Scholar] [CrossRef]
- Sunol, A.; Charan, S.; Seinfeld, J. Computational simulation of the dynamics of secondary organic aerosol formation in an environmental chamber. Aerosol Sci. Technol. 2018, 52, 470–482. [Google Scholar] [CrossRef]
- Karch, R.; Reischl, G.; Winkmayr, W.; Paulson, S.; Wang, S.; Yin, F.; Flagan, R.; Seinfeld, J. Smog Chamber Studies of Aerosol Formation in Atmospheric Mixtures. J. Aerosol Sci. 1989, 20, 995–998. [Google Scholar] [CrossRef]
- Jathar, S.; Donahue, N.; Adams, P.; Robinson, A. Testing secondary organic aerosol models using smog chamber data for complex precursor mixtures: Influence of precursor volatility and molecular structure. Atmos. Chem. Phys. 2014, 14, 5771–5780. [Google Scholar] [CrossRef]
- Carter, W.; Cocker, D., III; Fitz, D.; Malkina, I.; Bumiller, K.; Sauer, C.; Pisano, J.; Bufalino, C.; Song, C. A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation. Atmos. Environ. 2005, 39, 7768–7788. [Google Scholar] [CrossRef]
- Cocker, D., III; Flagan, R.; Seinfeld, J. State of the art chamber facility for studying atmospheric aerosol chemistry. Environ. Sci. Technol. 2001, 35, 2594–2601. [Google Scholar] [CrossRef]
- Ng, N.; Kwan, A.; Surratt, J.; Chan, A.; Chhabra, P.; Sorooshian, H.; Pye, J.; Crounse, P.; Wennberg, R.; Flagan, R.; et al. Secondary organic aerosol (SOA) formation from reaction of isoprene and nitrate radicals (NO3). Atmos. Chem. Phys. 2008, 8, 4117–4140. [Google Scholar] [CrossRef]
- Leone, J.; Flagan, R.; Grosjean, D.; Seinfeld, J. An Outdoor Smog Chamber and Modeling Study of Toluene-NOx Photooxidation. Int. J. Chem. Kinet. 1985, 17, 177–216. [Google Scholar] [CrossRef]
- Stanier, C.; Pathak, R.; Pandis, S. Measurements of volatility of aerosols from alpha-pinene ozonolysis. Environ. Sci. Technol. 2007, 41, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Donahue, N.; Shrivastava, M.; Weitkamp, E.; Sage, A.; Grieshop, A.; Lane, T.; Pierce, J.; Pandis, S. Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging. Science 2007, 315, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Mentel, T.; Bleilebens, D.; Wahner, A. A study of nighttime nitrogen oxidation oxidation in a large reaction chamber-the fate of NO2, N2O5, HNO3 and O3 at different humidities. Atmos. Environ. 1996, 30, 4007–4020. [Google Scholar] [CrossRef]
- Saathoff, H.; Naimann, K.-H.; Mohler, O.; Jonsson, A.; Hallquist, M.; Kiendler-Scharr, A. Temperature dependence of yields of secondary organic aerosols from the ozonolysis of a-pinene and limonene. Atmos. Chem. Phys. 2009, 9, 1551–1577. [Google Scholar] [CrossRef]
- Saathoff, H.; Moehler, O.; Schurath, O.; Kamm, S.; Dippel, B.; Mihekcic, L. The AIDA soot aerosol characterization campaign. J. Aerosol Sci. 2003, 34, 1277–1296. [Google Scholar] [CrossRef]
- Jonsson, A.; Hallquist, M.; Saathoff, H. Volatility of secondary aerosols from ozone-initiated oxidation of a-pinene and limonene. J. Aerosol Sci. 2007, 38, 843–852. [Google Scholar] [CrossRef]
- Rodenas, M.; Munoz, A.; EUPHORE Team. The EUPHORE chambers: A shared research platform to study atmospheric processes-20 years of international cooperation. In Proceedings of the EGU General Assembly, Vienna, Austria, 23–28 April 2017. [Google Scholar]
- Klotz, B.; Sorenson, S.; Barnes, L.; Becker, K.; Etzkorn, T.; Volkammer, R.; Platt, U.; Wirtz, K.; Martin-Reviejo, M. Atmospheric oxidation of toluene in a large-volume outdoor photoreactor: In situ determination of ring-retaining product yields. J. Phys. Chem. 1998, 102, 10289–10299. [Google Scholar] [CrossRef]
- Iinuma, Y.; Muller, C.; Berndt, T.; Boge, O.; Claeys, M.; Hermann, H. Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 6678–6683. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, D.; Dommen, J.; Kalberer, M.; Prevot, A.; Richter, R.; Sax, M.; Steinbacher, M.; Weingartner, E.; Baltensberger, U. Secondary Organic Aerosol Formation by Irradiation of 1,3,5-Trimethylbenzene-NOx-H2O in a New Reaction Chamber for Atmospheric Chemistry and Physics. Environ. Sci. Technol. 2005, 39, 668–2678. [Google Scholar] [CrossRef]
- Temime, B.; Healy, R.; Wenger, J. A denuder-filter sampling technique for the detection of gas and particle phase carbonyl compounds. Environ. Sci. Technol. 2007, 41, 6514–6520. [Google Scholar] [CrossRef]
- Healy, R.; Wenger, J.; Metzger, A.; Duplissy, J.; Kalberer, M.; Dommen, J. Gas/particle partitioning of carbonyls in the photooxidation of isoprene and 1, 3, 5-trimethylbenzene. Atmos. Chem. Phys. 2008, 8, 3215–3220. [Google Scholar] [CrossRef]
- Centre for Atmospheric Science. Smog Chamber Description. University of Manchester. Available online: www.manchester.ac.uk/restools/aerosolchamber/description (accessed on 14 April 2019).
- Song, C.; Na, K.; Cocker, D., III. Impact of the Hydrocarbon to NOx Ratio on Secondary Organic Aerosol Formation. Environ. Sci. Technol. 2005, 39, 31143–33149. [Google Scholar] [CrossRef]
- Tobias, M.; Ziemann, P. Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids and water with C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene. J. Phys. Chem. 2001, 105, 6129–6135. [Google Scholar] [CrossRef]
- Jeffries, H.; Fox, D.; Kamens, R. Outdoor Smog Chamber Studies (Effect of HC Reduction on Nitrogen Dioxide); EPA-650/3-75-011; Environmental Protection Agency: Research Triangle Park, NC, USA, 1975.
- Jeffries, H.; Kamens, R.; Sexton, K.; Gerhardt, A. Outdoor Smog Chamber Experiment to Test Photochemical Models; EPA Report 6003-82-016; Environmental Protection Agency: Research Triangle Park, NC, USA, 1982. [Google Scholar]
- Jeffries, H.; Kamens, R.; Sexton, K. Early history and rationale for outdoor chamber work at the University of North Carolina. Environ. Chem. 2012, 10, 349–364. [Google Scholar] [CrossRef]
- Lee, S.; Jang, M.; Kamens, R. SOA formation from the photooxidation of a-pinene in the presence of freshly emitted diesel soot exhaust. Atmos. Environ. 2004, 38, 2597–2605. [Google Scholar] [CrossRef]
- Edney, E.; Kleindienst, T.; Jaoui, M.; Lewandowski, M.; Offenberg, J.; Wang, J.; Claeys, M. Formation of 2-methyl tetrols, and 2-methylglyceric acid in secondary aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures an their detection in ambient PM2.5 samples collected in the eastern United States. Atmos. Environ. 2005, 39, 5281–5289. [Google Scholar] [CrossRef]
- Wiegand, A. Modelling Photochemical Production of Fine Particulates in a Toluene-NOx/Water Vapour System. Ph.D. Thesis, Queensland University of Technology, Brisbane, Australia, 1999. [Google Scholar]
- Hynes, R.; Angove, D.; Saunders, S.; Haved, H.; Azzi, M. Evaluation of two MCMv3.1 mechanisms using indoor environmental chamber data. Atmos. Environ. 2005, 39, 7251–7262. [Google Scholar] [CrossRef]
- Ren, Y.; Grosselin, B.; Daele, V.; Mellouki, A. Investigation of reaction of ozone with isoprene, methacrolein and methyl vinyl ketone using the HELIOS chamber. Faraday Discuss. 2017, 200, 289–311. [Google Scholar] [CrossRef]
- Sato, K.; Hatakeyama, S.; Imamura, T. Secondary organic aerosol formation during photooxidation of toluene: NOx dependence on chemical composition. J. Phys. Chem. 2007, 111, 9796–9808. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Z.; Hao, J.; Zhao, Z.; Li, J.; Hideto, T.; Hiroaki, T.; Akio, Y. Construction and characterization of an atmospheric simulation smog chamber. Adv. Atmos. Sci. 2007, 24, 250–258. [Google Scholar] [CrossRef]
- Hu, C.; Cheng, Y.; Pan, G.; Gai, Y.; Gu, X.; Zhao, W.; Wang, Z.; Zhang, W.; Chen, J.; Liu, F.; et al. A Smog Chamber Facility for Qualitative and Quantitative Study on Atmospheric Chemistry and Secondary Organic Aerosol. Chin. J. Chem. Phys. 2014, 6, 631–639. [Google Scholar] [CrossRef]
- Moon, K.-C.; Bae, G.-N.; Lee, S.-B.; Choi, J.-E. Some Study Using Twin Chambers Filled with Ambient Air; Air Resources Research Center, Korea Institute of Science and Technology: Seoul, Korea, 2019. [Google Scholar]
- Doussin, T.; Formenti, P.; Gratien, A.; Cirtog, M.; Cazaunau, M.; Michoud, V.; Baltensberger, U.; Prati, P.; Brotto, P.; Munoz, A.; et al. Eurochamp 2020. Available online: www.researchgate.net/project/EUROCHAMP2020 (accessed on 12 June 2019).
- Kaltsonoudis, C. A portable dual-smog-chamber system for atmospheric aerosol field studies. Atmos. Meas. Tech. 2019, 12, 2733–2743. [Google Scholar] [CrossRef]
- Miracolo, M.; Hennigan, C.; Raajan, M.; Gordon, N.; Lipsky, E.; Presto, A.; Donahue, N.; Robinson, A. Secondary aerosol formation from photochemical aging of aircraft exhaust in a smog chamber. Atmos. Chem. Phys. 2011, 11, 41356–41414. [Google Scholar] [CrossRef]
- Delaria, E.; Vieira, J.; Cremieux, J.; Cohen, R. Measurements of NO and NO2 exchange between the atmosphere and Quecus agrifolia. Atmos. Chem. Phys. 2008, 18, 14161–14173. [Google Scholar] [CrossRef]
- Mentel, T.; Wildt, J.; Kiendler-Scharr, A.; Kleist, E.; Tillmann, R.; Dal Maso, M.; Fisseha, R.; Hohas, T.; Spahn, H.; Uerlings, R.; et al. Photochemical production of aerosols from real plant emissions. Atmos. Chem. Phys. 2009, 9, 4387–4406. [Google Scholar] [CrossRef]
- Price, D. Field and Smog Chamber Studies of Agricultural Emissions and Reaction Products. Master’s Thesis, Utah State University, Logan, UT, USA, 2010. [Google Scholar]
- Baltensperger, U.; Dommen, J.; Alfarra, M.; Duplissy, J.; Gaeggeler, K.; Metzger, A.; Facchini, C.; Decesari, S.; Finessi, E.; Reinnig, C. Combined Determination of the Chemical Composition and Health Effects of Secondary Aerosols: The Polysoa Project. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, J.E.; Crosby, D.G.; Seiber, J.N. Vapor-phase photochemistry of pesticides. Residue Rev. 1983, 85, 1215–1224. [Google Scholar]
- Borras, E.; Rodenas, M.; Vasquesz, M.; Vera, T.; Munoz, A. Particulate and gas-phase products from the atmospheric degradation of chlorphyifos and chlorpyrifos-oxon. Atmos. Environ. 2015, 123A, 112–130. [Google Scholar] [CrossRef]
- de Gouw, J.; Warneke, C. Measurement of VOCs in the Earth’s Atmosphere using Proton Transfer Spectroscopy. Mass Spectr. Rev. 2007, 26, 223–257. [Google Scholar] [CrossRef]
- Kelly, T.; Fortune, C. Continous Monitoring of Gaseous Formaldehyde using an Improved Fluorescence Approach. Int. J. Environ. Anal. Chem. 1994, 54, 249–263. [Google Scholar] [CrossRef]
- Keffmann, J.; Heland, J.; Kurtencach, R.; Lorzer, J.; Wiesen, P. A new instrument (LOPAP) for detection of nitrous acid (HONO). Environ. Sci. Poll. Res. 2001, 9, 48–54. [Google Scholar]
- Crounse, J.; McKinney, K.; Kwan, A.; Wennberg, P. Measurements of gas-phase hydroperoxides by chemical ionization mass spectroscopy. Anal. Chem. 2006, 78, 6726–6732. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Noone, B.; O’Sullivan, D.; Heikes, B. Method for the Collection and HPLC Analysis of Hydrogen peroxide and C1 and C2 Hydroperoxides in the Atmosphere. J. Atmos. Ocean. Technol. 1995, 12, 1060–1070. [Google Scholar] [CrossRef]
- Hard, T.; George, L.; O’Brien, R. FAGE Determination of Tropospheric OH and HO2. J. Atmos. Sci. 1995, 52, 3354–3372. [Google Scholar] [CrossRef]
- Cocker, D., III; Whitlock, N.; Collins, D.; Wang, J.; Flagan, R.; Seinfeld, J. Instrumentation for state-of-the-art aerosol measurements in smog chambers. In Proceedings of the Combined US/German Ozone/Fine Particle Science and Environmental Chamber Workshop, Riverside, CA, USA, 4–6 October 1999. [Google Scholar]
- Mertes, P.; Pfaffenberger, L.; Dommen, J.; Kalberer, M.; Baltensperger, U. Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP). Atmos. Meas. Tech. 2012, 5, 2339–2348. [Google Scholar] [CrossRef]
- Baltensperger, U.; Chirico, R.; DeCarlo, P.; Dommen, J.; Gaeggeler, K.; Heringa, M.; Li, M.; Prevot, A.; Alfarra, M.; Gross, D.; et al. Recent developments in the mass spectroscopy of atmospheric aerosols. Eur. J. Mass Spectrom. 2010, 16, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bruins, A. Mass Spectroscopy with ion Sources Operating at Atmospheric Pressure. Mass Spectrom. Rev. 1991, 10, 53–77. [Google Scholar] [CrossRef]
- Fenn, J.; Mann, M.; Meng, C.; Wong, S.; Whitehouse, C. Electrostatic ionization for mass spectroscopy of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Lopez-Hilfiker, F.; Mohr, C.; Ehn, M.; Rubach, F.; Kleist, S.; Wildt, J.; Mentrel, T.; Lutz, A.; Hallquist, M.; Worsnop, D.; et al. A novel method for online analysis of gas and particle composition: Description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Atmos. Meas. Tech. 2014, 7, 983–1001. [Google Scholar] [CrossRef]
- Lee, B.; Lopez-Hilfiker, F.; Mohr, C.; Kurten, T.; Worsnop, D.; Thornton, J. An Iodide-Adduct High Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds. Environ. Sci. Technol. 2014, 48, 6309–6317. [Google Scholar] [CrossRef]
- Gross, D.; Galli, M.; Kalberer, M.; Prevot, A.; Dommen, J.; Duplissy, J.; Gascho, A.; Mertzger, A.; Alfarra, M.; Gaeggeler, K.; et al. Real time measurement of oligomeric species in secondary organic aerosol with the aerosol time-of-flight spectrometer. Anal. Chem. 2006, 78, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deming, B.; Sat, S.; Palm, B.; Talukder, R.; Roberts, J.; Veres, P.; Krechmer, J.; Thornton, A. Effects of Gas-Wall Interactions on Measurements of Semivolatile Compounds and Small Polar Compounds. Atmos. Atmos. Meas. Tech. 2019, 12, 3137–3149. [Google Scholar] [CrossRef]
- Pagonis, D.; Krechmer, J.; de Gouw, J.; Jiminez, J.; Ziermann, P. Effects of gas-wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds. Atmos. Meas. Tech. 2010, 10, 4687–4696. [Google Scholar] [CrossRef]
- Fuchs, H.; Ball, S.; Bohn, B.; Brauers, T.; Cohen, R.; Dorn, H.-P.; Dube, W.; Fry, J.; Haseler, R.; Heitmann, U.; et al. Intercomparison of measurements of NO2 concentrations in the atmospheric simulation chamber SAPHIR during the NO3Comp campaign. Atmos. Meas. Tech. 2010, 3, 21–37. [Google Scholar] [CrossRef]
- Fuchs, H.; Novelli, A.; Rolleter, M.; Hofzumahaus, A.; Pfannestill, E.; Kessel, S.; Edtbauer, A.; Willams, J.; Michoud, V.; Dusnter, S.; et al. Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR. Atmos. Meas. Tech. 2017, 10, 4023–4053. [Google Scholar] [CrossRef]
- Onel, L.; Brennan, A.; Gianella, M.; Ronnie, G.; Lawry Aguilla, A.; Hancock, G.; Whalley, L.; Seakins, P.; Richie, G.; Heard, D. An intercomparison of HO2 measurements by fluorescence assay by gas expansion and cavity ring down spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry). Atmos. Meas. Tech. 2017, 10, 4877–4894. [Google Scholar] [CrossRef]
- Jeffries, H.; Sexton, K.; Yu, J. Atmospheric Photochemistry Studies of Pollutant Emissions from Transportation Vehicles Operating on Alternative Fuels; Final Report; National Renewable Energy Laboratory: Golden, CO, USA, 1998.
- Wiesen, P. Investigation of Real Car Exhaust in the EURPHORE Chamber; Combined US/German Ozone/Fine Particle Science and Environmental Chamber Workshop: Riverside, CA, USA, 1999. [Google Scholar]
- Geiger, H.; Kleffmenn, J.; Wiesen, P. Smog chamber studies on the influence of diesel exhaust on photosmog formation. Atmos. Environ. 2002, 36, 1737–1747. [Google Scholar] [CrossRef]
- Chirico, R.; De Carlo, P.; Heringa, M.; Tritscher, T.; Richter, A.; Pevot, A.; Dommen, J.; Weungartne, E.; Wehrle, G.; Gysel, M.; et al. Impact of After treatment Devices on Primary Emissions and Secondary Organic Aerosol Formation Potential from in-Use Diesel Vehicles: Results from Smog Chamber Experiments. Atmos. Chem. Phys. 2010, 10, 11545–11563. [Google Scholar] [CrossRef]
- Zielinska, B.; Samy, S.; Seagrave, J.; Mc Donald, J.; Wirtz, K.; Vazquez, M. Investigation of atmospheric transformations of diesel emissions in the European Photoreactor (EURPHORE). In Proceedings of the 5th Asian Aerosol Conference, Kaohsinung, Taiwan, 26–29 August 2007. [Google Scholar]
- Weitkamp, E.; Sage, A.; Pierce, J.; Donahue, N.; Robinson, A. Organic Aerosol Formation from Photochemical Oxidation of Diesel Exhaust in a Smog Chamber. Environ. Sci. Technol. 2007, 41, 6969–6975. [Google Scholar] [CrossRef]
- Jathar, S.; Gordon, T.; Hennigan, C.; Pye, H.; Pouliot, G.; Adams, P.; Donahue, N.; Robinson, A. Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States. Proc. Natl. Acad. Sci. USA 2014, 111, 10473–10478. [Google Scholar] [CrossRef]
- Bruns, E.; Krapf, M.; Orasche, J.; Huang, Y.; Zimmeman, R.; Drinovec, L.; Mocnik, G.; El-Haddad, I.; Slowik, J.; Dommen, J.; et al. Characterization of primary and secondary wood combustion products generated from different burner loads. Atmos. Chem. Phys. 2015, 15, 2825–2841. [Google Scholar] [CrossRef]
- Tkacik, D.; Robinson, E.; Ahern, A.; Sale, R.; Stockwell, C.; Veres, P.; Simpson, I.; Meinardi, S.; Blake, D.; Yokelson, R.; et al. A dual-chamber for quantifying the effects of atmospheric perturbations on secondary organic aerosol formation from biomass burning emissions. J. Geophys. Res. Atmos. 2017, 122, 6043–6058. [Google Scholar] [CrossRef]
- Graedel, T.; Wechsler, C. Chemistry within aqueous atmospheric aerosols and raindrops. Rev. Geophys. 1981, 19, 505–539. [Google Scholar] [CrossRef]
- George, I.; Abbatt, J. Heterogeneous oxidation of atmospheric aerosol particles by gas phase radicals. Nat. Chem. 2010, 2, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Carter, W. Kinetics and mechanisms of the gas-phase reactions of ozone with organic compounds under atmospheric conditions. Chem. Rev. 1984, 84, 437–470. [Google Scholar] [CrossRef]
- Calvert, J.; Orlando, J.; Stockwell, W.; Wallington, T. The Mechanisms of Reactions Influencing Atmospheric Ozone; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- International Union of Pure and Applied Chemistry (IUPAC). Task Group on Atmospheric Chemical Kinetic Data Evaluation; Evaluated Data Sheets; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2019. [Google Scholar]
- Carter, W. A detailed mechanism for the gas-phase atmospheric reactions of organics. Atmos. Environ. 1990, 24, 481–518. [Google Scholar] [CrossRef]
- Carter, W. Development of the SAPRC-07 chemical mechanism. Atmos. Environ. 2010, 44, 5324–5335. [Google Scholar] [CrossRef]
- Jenkin, M.; Saunders, D.; Wagner, V.; Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v3, (Part B): Tropospheric degradation of aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 181–193. [Google Scholar] [CrossRef]
- Saunders, S.; Jenkin, M.; Derwent, R.; Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v.3 (Part A): Tropospheric degradation of non-aromatic organic compound. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef]
- Kerr, J.A.; Calvert, J. Chemical Transformation Modules for Eulerian Acid Deposition Models: Volume 2 The Gas Phase Chemistry; EPA Report 600/S3-85/015; US Environmental Protection Agency: Research Triangle Park, NC, USA, 1985.
- Stockwell, W.; Middleton, P.; Chang, J. The Second Generation Regional Acid Deposition Model Chemical Mechanism for Regional Air Quality Modeling. J. Geophys. Res. 1990, 95, 16343–16367. [Google Scholar] [CrossRef]
- Whitten, G.; Hogo, H.; Killus, J. The carbon-bond mechanism: A condensed kinetic mechanism for photochemical smog. Environ. Sci. Technol. 1980, 14, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Gery, M.W.; Whitten, G.Z.; Killus, J.P.; Dodge, M.C. A photochemical kinetics mechanism for urban and regional scale computer modeling. J. Geophys. Res. Space Phys. 1989, 94, 12925. [Google Scholar] [CrossRef]
- Yarwood, G.; Rao, S.; Yocke, M.; Whitten, G. Updates to the Carbon Bond Chemical Mechanism: CB05; Final Report to US; RT-0400675; Environmental Protection Agency: Washington, DC, USA, 2005. [Google Scholar]
- Goliff, W.; Stockwell, W. The Regional Atmospheric Chemistry Mechanism, version 2; University of California: Davis, CA, USA, 2008. [Google Scholar]
- Dodge, M. Combined use of modeling techniques and smog chamber data to derive ozone-precursor relationships. In Proceedings of the International Conference on Photochemical Oxidant Pollution and Its Control, Raleigh, NC, USA, 12–17 September 1976; EPA Report 600/3-77-001b. US Environmental Protection Agency: Research Triangle Park, NC, USA, 1977; pp. 881–889. [Google Scholar]
- National Academy Sciences (US). The Relationship of Emissions to Ambient Air Quality. Report of the Coordinating Committee on Air Quality Studies; Committee of Public Works, US Senate: Washington, DC, USA, 1974.
- Seigneur, C.; Dennis, R. Atmospheric Modeling. In Technical Challenges of Multipollutant Air Quality Management; Hidy, G., Brook, J., Demerjian, K., Molina, L., Pennell, W., Scheffe, R., Eds.; Springer: New York, NY, USA, 2011; pp. 299–339. [Google Scholar]
- Reynolds, S.; Roth, P.; Seinfeld, J. Mathematical modeling of photochemical air pollution-I. Formulation of the model. Atmos. Environ. 1973, 7, 1033–1061. [Google Scholar] [CrossRef]
- Chang, J.; Middleton, P.; Stockwell, W.; Walcek, C.; Pleim, J.; Lansford, H.; Binkowski, F.; Madronich, S.; Seaman, N.; Stauffer, D.; et al. The Regional Acid Deposition Model and Engineering Model. State-of-Science/Technology; Report 4; National Precipitation Assessment Program: Washington, DC, USA, 1991.
- Binkowski, F.; Roselle, S. Models 3-Community Multiscale Air Quality Model (CMAQ) model aerosol component 1. Model description. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Simonaitis, J.; Meagher, J.; Bailey, E. Evaluation of the condensed carbon bond (CB-IV) mechanism against smog chamber data at low VOC and NOx concentrations. Atmos. Environ. 1997, 31, 27–43. [Google Scholar] [CrossRef]
- Bloss, C.; Wagner, V.; Bonzanini, A.; Jenkin, M.; Wirtz, K.; Martin-Reviejo, M.; Pilling, M. Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data. Atmos. Chem. Phys. 2005, 5, 623–639. [Google Scholar] [CrossRef]
- Parikh, M.; Jeffries, H.; Sexton, K.; Luecken, D.; Kamens, R.; Vizuete, W. Evaluation of Aromatic Oxidation Reactions in Seven Chemical Mechanisms with an Outdoor Chamber. Environ. Chem. 2013, 10, 245–259. [Google Scholar] [CrossRef]
- Carter, W.; Lurmann, F. Evaluation of a detailed gas-phase atmospheric reaction mechanism using environmental chamber data. Atmos. Environ. 1991, 25, 2771–2806. [Google Scholar] [CrossRef]
- Carter, W. Status of Research on VOC Reactivity in the United States. In Proceedings of the 1st US-German Workshop on the Photochemical Ozone Problem and its Control, Berlin, Germany, 24–27 September 1996. [Google Scholar]
- Farmer, D.; Perring, A.; Wooldridge, P.; Blake, D.; Baker, A.; Meinardi, S.; Huey, L.; Tanner, D.; Vargas, O.; Cohen, R. Impact of organic nitrates on urban ozone production. Atmos. Chem. Phys. 2011, 11, 4085–4094. [Google Scholar] [CrossRef]
- Spicer, C.; Miller, D. Nitrogen Balance in Smog Chamber Studies. J. Air Pollut. Control Assoc. 1976, 26, 45–50. [Google Scholar] [CrossRef]
- Hidy, G.M.; Burton, C.S. Atmospheric Aerosol Formation by Chemical Reactions. In The Character and Origins of Smog Aerosols; Hidy, G.M., Mueller, P., Grosjean, D., Appel, B., Wesolowski, J., Eds.; Wiley-Interscience: New York, NY, USA, 1980; pp. 385–432. [Google Scholar]
- Grosjean, D.; Seinfeld, J. Parameterization of the Formation Potential of Secondary Organic Aerosols. Atmos. Environ. 1989, 23, 1733–1747. [Google Scholar] [CrossRef]
- Odum, J.; Hofmann, T.; Bowman, F.; Collins, P.; Flagan, R.; Seinfeld, J. Gas Particle Partitioning and Secondary Organic Aerosol Yields. Environ. Sci. Technol. 1996, 30, 2580–2585. [Google Scholar] [CrossRef]
- Ng, N.; Kroll, J.; Chan, A.; Chabra, P.; Flagan, R.; Seinfeld, J. Secondary aerosol formation from m-xylene, toluene and benzene. Atmos. Chem. Phys. 2007, 7, 3909–3922. [Google Scholar] [CrossRef]
- Nolting, F.; Behnke, W.; Zetzsch, C. A smog chamber for studies of the reactions of terpenes and alkanes with ozone and OH. J. Atmos. Chem. 1988, 6, 47–59. [Google Scholar] [CrossRef]
- Hu, D.; Tolocka, M.; Li, Q.; Kamens, R. A kinetic mechanism for predicting secondary organic aerosol formation from toluene oxidation in the presence of NOx and natural sunlight. Atmos. Environ. 2007, 41, 6478–6496. [Google Scholar] [CrossRef]
- Matsunaga, A.; Docherty, K.; Lim, Y.; Ziemann, P. Composition and yields of secondary organic aerosol formed from OH radical-initiated reactions of linear alkenes in the presence of NOx: Modeling and measurements. Atmos. Environ. 2009, 43, 1348–1357. [Google Scholar] [CrossRef]
- Blanchard, C.; Hidy, G.; Tanenbaum, S.; Edgerton, E.; Hartsell, B.; Jansen, J. Carbon in southeastern U.S. aerosol particles: Empirical estimates of secondary organic aerosol formation. Atmos. Environ. 2008, 42, 6710–6720. [Google Scholar] [CrossRef]
- Herndon, S.; Onasch, T.; Wood, E.; Kroll, J.; Canagaratna, M.; Jayne, J.; Zavala, M.; Knighton, W.B.; Mazzoleni, C.; Dubey, M.; et al. Correlation of secondary organic aerosol with odd oxygen in Mexico City. Geophys. Res. Lett. 2008, 35, L15804. [Google Scholar] [CrossRef]
- O’Neal, H.E.; Blumstein, C. A new mechanism for gas phase ozone-olefin reactions. Int. J. Chem. Kinet. 1973, 5, 397–413. [Google Scholar] [CrossRef]
- McMurry, P.; Shepherd, M.; Vickery, J. (Eds.) Particulate Matter Science for Policy Makers; Cambridge University Press: Cambridge, UK, 2004; p. 22. [Google Scholar]
- Pankow, J.; Asher, W. SIMPOL.1: A simple contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. 2008, 8, 2773–2796. [Google Scholar] [CrossRef]
- Cocker, D., III; Li, W.; Li, L.; Kacarab, M.; Peng, W. Air Quality Impacts of Low Vapor Pressure-Volatile Organic Compounds; Contract No. 13-302; Final Report; California Air Resources Board: Sacramento, CA, USA, 2016.
- Kroll, J.; Ng, N.; Murphy, S.; Flagan, R.; Seinfeld, J. Secondary organic aerosol formation from isoprene photooxidation. Environ. Sci. Technol. 2006, 49, 1869–1877. [Google Scholar] [CrossRef]
- Jang, M.; Czoschke, N.M.; Lee, S.; Kamens, R.M. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 2002, 298, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Wang, Z.; Huang, M.; Fang, L.; Zhang, W. Effects of seed aerosols on the growth of secondary organic aerosols from the photooxidation of toluene. J. Environ. Sci. 2007, 19, 704–708. [Google Scholar] [CrossRef]
- Chu, B.; Jiang, J.; Wang, K.; Li, J.; Hao, J. Effects of Inorganic Seeds on Secondary Organic Aerosol (SOA) Formation. In Atmospheric Aerosols-Regional Characteristics-Chemistry and Physics; IntechOpen: London, UK, 2012. [Google Scholar]
- Chu, B.; Liu, T.; Zhang, X.; Liu, Y.; Ma, Q.; Ma, J.; He, H.; Wang, X.; Li, J.; Hao, J. Secondary aerosol formation and oxidation capacity in photooxidation in the presence of Al2O3 seed particles and SO2. Sci. China Chem. 2015, 58, 1426–1434. [Google Scholar] [CrossRef]
- Kleindienst, T.E.; Edney, E.O.; Lewandowski, M.; Offenberg, J.; Jaoui, M. Secondary Organic Carbon and Aerosol Yields from the Irradiations of Isoprene and a-Pinene in the Presence of NOx and SO2. l. Environ. Sci. Technol. 2006, 40, 3807–3812. [Google Scholar] [CrossRef]
- Donahue, N.; Robinson, A.; Stanier, C.; Pandis, S. Coupled partitioning, dilution, and chemical aging of semi-volatile organics. Environ. Sci. Technol. 2006, 40, 2635–2643. [Google Scholar] [CrossRef]
- 201. Paulot, F.; Crounse, J.; Kjaergaard, H.; Kurten, A.; St. Clair, J.; Seinfeld, J.; Wennberg, P. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science 2009, 325, 730–733. [Google Scholar] [CrossRef]
- Surratt, J.; Chan, A.; Eddingsaas, N.; Chan, M.-N.; Loza, C.; Kwan, A.; Hersey, S.; Flagan, R.; Wennberg, P.; Seinfeld, J. Reactive Intermediates Revealed in Secondary Organic Aerosol Formation from Isoprene. Proc. Natl. Acad. Sci. USA 2010, 107, 6640–6645. [Google Scholar] [CrossRef]
- Lopez-Hilfiker, F.; Mohr, C.; D’Ambro, E.; Lutz, A.; Riedel, T. Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA. Environ. Sci. Technol. 2016, 50, 2200–2209. [Google Scholar] [CrossRef]
- Kalberer, M.; Paulsen, D.; Sax, M.; Steinbacher, J.; Dommen, J.; Prevot, A.; Fisseha, R.; Weingartner, E.; Frankevich, V.; Zenobi, R.; et al. Identification of polymers as major components of atmospheric organic aerosols. Science 2004, 303, 1659–1662. [Google Scholar] [CrossRef]
- Isaacman-Van Wertz, G.; Massoli, P.; O’Brien, R.; Lim, C.; Franklin, J.; Moss, J.; Hunter, J.; Nowak, J.; Canagaratna, M.; Misztal, P.; et al. Chemical evolution of atmospheric organic carbon over multiple generations of oxidation. Nat. Chem. 2018, 10, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Donahue, N.; Henry, K.; Mentel, T.; Kiendler-Scharr, A.; Spindler, C.; Bohn, B.; Brauers, T.; Dorn, H.; Fuchs, H.; Tillman, R.; et al. Aging of biogenic secondary organic aerosol for gas-phase radical reactions. Proc. Natl. Acad. Sci. USA 2012, 109, 13503–13508. [Google Scholar] [CrossRef] [PubMed]
- Donahue, N.; Epstein, S.; Pandis, S.; Robinson, A. A two-dimensional volatility basis set: Organic-aerosol mixing thermodynamics. Atmos. Chem. Phys. 2011, 11, 3303–3318. [Google Scholar] [CrossRef]
- Chacon-Madrid, H.; Murphy, B.; Pandis, S.; Donahue, N. Simulations of Smog-Chamber Experiments Using the Two-Dimensional Volatility Basis Set: Linear Oxygenated Precursors. Environ. Sci. Technol. 2012, 46, 11179–11186. [Google Scholar] [CrossRef] [PubMed]
- Martin, L. Kinetic Studies of Sulfite Oxidation in Aqueous Solutions. Acid Precipitation: SO2, NO, NO2 Oxidation Mechanisms: Atmospheric Conditions; Calvert, J., Ed.; Ann Arbor Scientific Publications: Ann Arbor, MI, USA, 1984; pp. 63–100. [Google Scholar]
- Nenes, A.; Pandis, S.; Pilinis, C. ISORROPIA: A new thermodynamic model for multiphase multicomponent inorganic aerosols. Aquat. Geochem. 1998, 4, 123–152. [Google Scholar] [CrossRef]
- Altieri, K.; Carlton, A.; Turpin, B.; Seitzinger, S. Formation of Oligomers in Cloud-Processing Reactions of Isoprene Oxidation Products. Environ. Sci. Technol. 2006, 40, 4956–4960. [Google Scholar] [CrossRef]
- Ervens, B.; Turpin, B.; Weber, R. Secondary organic aerosol formation in cloud droplets and aqueous paricles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102. [Google Scholar] [CrossRef]
- Romonosky, D.; Li, Y.; Shiraiwa, M.; Laskin, A.; Laskin, J.; Nizkorodov, S. Aqueous Photochemistry of Secondary Aerosol of a-Pinene and a-Humulene Oxidized with Ozone, Hydroxyl Radical, and Nitrate Radical. J. Phys. Chem. 2017, 121, 1298–1309. [Google Scholar] [CrossRef]
- Hinks, M.; Montoya-Aguilera, J.; Ellison, L.; Lin, P.; Laskin, A.; Laskin, J.; Shiraiwa, M.; Dabdub, D.; Nizkorodov, S. Effect of relative humidity on the composition of secondary organic aerosol formed from the oxidation of toluene. Atmos. Chem. Phys. 2018, 18, 1643–1652. [Google Scholar] [CrossRef]
- Tillmann, R.; Hallquist, M.; Jonsson, A.; Kiendler-Scherr, A.; Saathoff, H.; Ianuma, Y.; Mentel, T. Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of a-pinene. Atmos. Chem. Phys. 2010, 10, 7057–7072. [Google Scholar] [CrossRef]
- Saxena, P.; Hildemann, L.M.; McMurry, P.; Seinfeld, J. Organics alter hygroscopic behavior of atmospheric particles. J. Geophys. Res. 1995, 100, 18753–18770. [Google Scholar] [CrossRef]
- Folkers, M.; Menel, T.; Wahner, A. Influence of an organic coating on the reactivity of aqueous aerosols probed by heterogeneous hydrolysis of N2O. Geophys. Res. Lett. 2003, 30, 1644. [Google Scholar] [CrossRef]
- Ziemann, P.; Atkinson, R. Kinetics, products and mechanisms of secondary aerosol formation. Chem. Soc. Rev. 2012, 41, 6582–6605. [Google Scholar] [CrossRef]
- Middleton, P.; Brock, J. Simulation of aerosol kinetics. J. Colloid Interface Sci. 1976, 54, 249–264. [Google Scholar] [CrossRef]
- Griffin, R.; Dabdub, D.; Seinfeld, J. Secondary organic aerosol 1. Atmospheric chemical mechanism for production of molecular constituents. J. Geophys. Res. Atmos. 2002, 107, 3–26. [Google Scholar] [CrossRef]
- Pun, B.; Griffin, R.; Seigneur, C.; Seinfeld, J. Secondary organic aerosol 2. Thermodynamic model for gas/particle partitioning of molecular constituents. J. Geophys. Res. Atmos. 2002, 107, 4333. [Google Scholar] [CrossRef]
- Chan, A.; Kroll, J.; Ng, N.; Seinfeld, J. Kinetic modeling of secondary organic aerosol formation: Effects of particle-and gas-phase reactions of semivolatile products. Atmos. Chem. Phys. 2007, 7, 4135–4147. [Google Scholar] [CrossRef]
- Li, J.; Cleveland, M.; Ziemba, L.; Griffin, E.; Barsanti, K.; Pankow, J.; Ying, Q. Modeling Regional Secondary Organic Aerosol Using the Master Chemical Mechanism. Atmos. Environ. 2015, 102, 52–61. [Google Scholar] [CrossRef]
- Jathar, S.; Cappa, C.; Wexler, A.; Seinfeld, J.; Kleeman, M. Multidimensional oxidation model to simulate secondary organic aerosol in a 3-D air quality model. Geosci. Model Dev. 2015, 8, 2553–2567. [Google Scholar] [CrossRef]
- US Environmental Protection Agency. Mechanisms in the CMAQv5.1 Model. Available online: www.airquality modeling.org/index.php/CMAQv5.1_Mechanisms (accessed on 5 June 2019).
- Vernecek, M.; Cai, C.; Kaduwela, A.; Avise, J.; Carter, W.; Kleeman, M. Analysis of SAPRC16 chemical mechanism for ambient simulations. Atmos. Environ. 2018, 192, 136–150. [Google Scholar] [CrossRef]
- National Center for Atmospheric Research (NCAR). Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A). Available online: www2.acom.ucar.edu/modeling/ecko (accessed on 6 June 2019).
- Lannuque, V.; Camredon, M.; Couvidat, F.; Hodzic, A.; Valorso, R.; Madronich, S.; Bessagnet, B.; Aumont, B. Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: Development of the VBS-GECKO parameterization. Atmos. Chem. Phys. Discuss. 2018, 18, 13411–13428. [Google Scholar] [CrossRef]
- Praske, E.; Otkjaar, R.; Croiunse, J.; Hethcox, J.C.; Stoltz, B.; Kjaergaard, H. Atmospheric autoxidation is increasingly important in urban and suburban North America. Proc. Natl. Acad. Sci. USA 2018, 115, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Coudivat, F.; Sartelet, K.; Seigneur, C. Comparison of Different Gas-Phase Mechanisms and Aerosol Modules for Simulating Particulate Matter Formation. J. Air Waste Manag. Assoc. 2011, 61, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Stanier, C.O.; Donahue, N.M.; Pandis, S.N. Parameterization of secondary organic aerosol mass fractions from smog chamber data. Atmos. Environ. 2008, 42, 2276–2299. [Google Scholar] [CrossRef]
- Hidy, G.M. Worldwide aerosol chemistry: From hemispheric distributions to megacity sources. J. Air Waste Manag. Assoc. 2009, 59, 770–789. [Google Scholar]
- Jimenez, M.; Canagarata, N.; Donahue, N.; Prevot, A.; Zhang, Q.; Kroll, J.; DeCarlo, P.; Allan, J.; Coe, H.; Ng, N.; et al. Evolution of Organic Aerosols in the Atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.; Huntzicker, J. Identification of Secondary Organic Aerosol Episodes and Quantification of Primary and Secondary Organic Aerosol Concentrations during SCAQS. Atmos. Environ. 1995, 29, 327–354. [Google Scholar] [CrossRef]
- Zhang, Q.; Alfarra, M.; Worsnop, D.; Allan, J.; Coe, H.; Canagarata, M.; Jiminez, J. Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry. Environ. Sci. Technol. 2005, 39, 4938–4952. [Google Scholar] [CrossRef]
- Rinaldi, M.; Gilardoni, S.; Paglione, M.; Sandrini, S.; Fuzzi, S.; Massoli, P.; Bonasoni, P.; Cristofanelli, P.; Marinoni, A.; Poluzzi, V.; et al. Organic aerosol evolution and transport observed at Mt. Cimone (2165 m asl), Italy, during the PEGASOS campaign. Atmos. Chem. Phys. 2015, 15, 11327–11340. [Google Scholar] [CrossRef]
- Carlton, A.; DeGouw, J.; Jiminez, J.; Ambrose, J.; Attwood, A.; Brown, S.; Baker, K.; Brock, C.; Cohen, R.; Edgerton, S.; et al. Synthesis of the Southeast Atmosphere Studies. Bull. Am. Meteorol. Soc. 2018, 98, 547–567. [Google Scholar] [CrossRef]
- Mao, J.; Carlton, A.; Cohen, R.; Brune, W.; Brown, S.; Wolfe, G.; Jiminez, J.; Pye, H.; Lee Ng, N.; Xu, L.; et al. Southeast Atmosphere Studies: Learning from model-observation syntheses. Atmos. Chem. Phys. 2018, 18, 2615–2651. [Google Scholar] [CrossRef]
- Kourtchev, I.; Ruukanen, T.; Maenhaut, W.; Kulmala, M.; Claeys, M. Observations of 2-methyltetrols and related photooxidation products of isoprene in boreal forest aerosols from Hyytiala, Finland. Atmos. Chem. Phys. 2005, 5, 2761–2770. [Google Scholar] [CrossRef]
- Froyd, K.; Murphy, S.; Murphy, D.; de Gouw, J.; Eddingrass, N.; Wennberg, P. Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass. Proc. Natl. Acad. Sci. USA 2010, 107, 21360–21365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Worton, D.R.; Lewandowski, M.; Ortega, J.; Rubitschun, C.L.; Park, J.-H.; Kristensen, K.; Campuzano-Jost, P.; Day, D.A.; Jiménez, J.L.; et al. Organosulfates as Tracers for Secondary Organic Aerosol (SOA) Formation from 2-Methyl-3-Buten-2-ol (MBO) in the Atmosphere. Environ. Sci. Technol. 2012, 46, 9437–9446. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Reinnig, M.-C.; Naumann, K.; Saathoff, H.; Menetel, T.; Donahue, N.; Hoffmann, Y. Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid-a mass spectrometric study of SOA aging. Atmos. Chem. Phys. 2012, 12, 1483–1496. [Google Scholar] [CrossRef]
- Zheng, M.; Cass, G.; Schauer, J.; Edgerton, E. Source apportionment of PM2.5 in the southeastern United States using solvent extractable organic compounds. Environ. Sci. Technol. 2001, 36, 2361–2371. [Google Scholar] [CrossRef]
- Eschenroeder, A.; Deley, G.; Wahl, R. Field Program Designs for Verifying Photochemical Models; Report EPA R4-73-012c; Environmental Protection Agency: Research Triangle Park, NC, USA, 1973.
- Fiegley, C.; Jeffries, H.; Kamens, R. An Experimental Simulation of Los Angeles Reactive Pollutant Program (LARPP) Operation 33—Part 1. Experimental Simulation in an Outdoor Smog Chamber. Atmos. Environ. 1979, 16, 1989–1996. [Google Scholar]
- Fiegley, C.; Jeffries, H. Analysis of Processes Affecting Oxidant and Precursors in the Los Angeles Reactive Pollutant Program (LARPP) Operation 33. Atmos. Environ. 1979, 13, 1369–1384. [Google Scholar] [CrossRef]
- Hidy, G.M. An Historical Experiment: Los Angeles Smog Evolution Observed by Blimp. J. Air Waste Manag. Assoc. 2018, 68, 634–655. [Google Scholar] [CrossRef]
- Edinger, J. Vertical Distribution of Photochemical Smog in Los Angeles Basin. Environ. Sci. Technol. 1973, 7, 247–252. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency (EPA). Community Multiscale Air Quality Modeling System (CMAQ). Available online: www.epa.gov/cmaq (accessed on 12 April 2019).
- Wang, M.; Penner, J.; Liu, X. Coupled IMPACT aerosol and NCAR CAM3 model: Evaluation of predicted aerosol number and size distribution. J. Geophys. Res. Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Kaiser, J.C.; Hendricks, J.; Righi, M.; Jöckel, P.; Tost, H.; Kandler, K.; Weinzierl, B.; Sauer, D.; Heimerl, K.; Schwarz, J.P.; et al. Global aerosol modeling with MADE-3 (v3.0) in EMAC (based on v2.53): Model description and evaluation. Geosci. Model Dev. 2019, 12, 541–579. [Google Scholar]
- Long, M.; Yantosca, J.; Nielsen, J.; Keller, C.; da Silva, M.; Pawson, S.; Jacob, D. Development of a Grid-independent GEOS-Chem Chemical Transport Model as an Atmospheric Chemistry Module or Earth System Models. Geosci. Model Dev. 2014, 7, 7505–7524. [Google Scholar] [CrossRef]
- Solomon, P.; Cowling, E.; Hidy, G.; Furiness, C. Comparison of scientific findings from major ozone field studies and aerosol precursors. Atmos. Environ. 2000, 34, 1885–1920. [Google Scholar] [CrossRef]
- Hidy, G.M. Atmospheric Sulfur and Nitrogen Oxides; Academic Press: San Diego, CA, USA, 1994. [Google Scholar]
- Hansen, D.A.; Edgerton, E.S.; Hartsell, B.; Jansen, J.; Hidy, G.; Kandaswamy, K.; Blanchard, C. The Southeastern Aerosol Research and Characterization study (SEARCH): 1. Overview. J. Air Waste Manag. Assoc. 2003, 53, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Solomon, P.; Sioutas, C. Continuous and semicontinuous monitoring techniques for particulate matter mass and chemical components: A synthesis of findings from EPA’s particulate matter supersites program and related studies. J. Air Waste Manag. Assoc. 2008, 58, 164–195. [Google Scholar] [CrossRef] [PubMed]
- Woody, M.C.; Baker, K.R.; Hayes, P.L.; Jimenez, J.L.; Koo, B.; Pye, H.O. Understanding sources of organic aerosol during CalNex-2010 using the CMAQ-VBS. Atmos. Chem. Phys. 2016, 16, 4081–4100. [Google Scholar] [CrossRef]
- Baker, K.; Kleindienst, T.; Offenberg, J.; Beaver, M.; Kelly, J.; Carlton, A. Evaluation of CMAQ Estimated Gas and Aerosol Carbon Using STN, Improve and CalNex Field Measurements. In Proceedings of the 13th Annual CMAS Conference, Research Triangle Park, NC, USA, 27–29 October 2014. [Google Scholar]
- Mebust, M.; Eder, B.; Binkowski, F.; Roselle, S. Models-3 Community Multiscale Air Quality (CMAQ) model aerosol component 2. Model Evaluation. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Ciarelli, C.; Aksoyoglu, S.; Crippa, M.; Jiminez, J.; Nemitz, E. Evaluation of European air quality modelled by CAMx including the volatility basis set scheme. Atmos. Chem. Phys. 2016, 16, 10313–10332. [Google Scholar] [CrossRef]
- Mircea, M.; Bessagnet, B.; D’Isidoro, M.; Pirovano, G.; Aksoyoglu, S.; Ciarelli, G.; Tsyro, S.; Manders, A.; Bieser, J.; Stern, R.; et al. EURODELTA III exercise: An evaluation of air quality models’ capacity to reproduce the carbonaceous aerosol. Atmos. Environ. X 2019, 2, 100018. [Google Scholar] [CrossRef]
- New York Deptartment of Environmental Conservation. CMAQ Model Performance Assessment 8-Hr OTC Ozone Modeling; Report TSD-1e; Bureau of Air Quality and Research Division: Albany, NY, USA, 2006.
- Appel, K.; Chemel, C.; Roselle, S.; Frabcis, X.; Hu, T.-M.; Sokhl, R.; Rao, S.; Galmarini, S. Examination of Community Multiscale Air Quality (CMAQ) model performance over the North American and European domains. Atmos. Environ. 2012, 53, 142–155. [Google Scholar] [CrossRef]
- Volkamer, R.; Jiminez, J.; San Martini, F.; Dzepina, K.; Salcedo, D.; Molina, L.; Worsnop, S.; Molina, M. Secondary organic aerosol formation from anthropogenic in pollution: Rapid and higher than expected. Geophys. Lett. 2006, 33, L17811. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Crounse, J.D.; Schwantes, R.H.; Teng, A.; Bates, K.H.; Zhang, X.; Clair, J.M.S.; Brune, W.H.; Tyndall, G.S.; Keutsch, F.N.; et al. Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): Mechanistic chamber studies on the oxidation of biogenic compounds. Atmos. Chem. Phys. 2014, 14, 13531–13549. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Review | Year | Content |
|---|---|---|
| Finlayson-Pitts and Pitts [28] | 2000 | Textbook-comprehensive review of gas and aerosol chemistry relevant to the atmosphere through the 1990s. |
| Dodge [36] | 2000 | Atmospheric chemistry of O3 with analysis of smog chambers and their results. |
| Atkinson [38] | 2000 | Photochemical mechanism review with relevance to atmospheric chemistry. |
| George et al. [39] | 2005 | Comprehensive review of heterogeneous atmospheric chemistry |
| Seinfeld and Pandis (3rd ed.) [29] | 2006 | Textbook-comprehensive gas and aerosol chemistry with summary of meteorological and climate relevance through early 2000s. |
| Kroll and Seinfeld [40] | 2008 | Review of photochemical aerosol formation with reference to smog chamber studies through mid- 2000s. |
| Hallquist et al. [37] | 2009 | Review of secondary organic aerosol (SOA) formation including photochemical processes. |
| Carlton et al., [41] | 2009 | Secondary organic aerosols (SOA) from isoprene oxidation. |
| Lim et al. [42] | 2010 | Aqueous chemistry and formation of SOA. |
| Stockwell et al. [43] | 2012 | Chemical mechanisms and models for O3. |
| Glasius and Goldstein [44] | 2016 | Review of contemporary atmospheric organic chemistry status and future emphasizing SOA. |
| Bianchi et al. [45] | 2019 | Review of autoxidation and highly oxygenated organic molecules (HOM) in atmospheric chemistry. |
| Location | Type | Volume (m3) | Material | Temperature (°K) | Reference a |
|---|---|---|---|---|---|
| California Inst. of Technology | Indoor photoreactor black light | 28 (dual) | Teflon (FEP) | 290–303 | Cocker [86]; Ng [87] |
| California Inst. of Technology | Outdoor photoreactor | 65 | FEP | ambient | Leone et al. [88] |
| Carnegie Mellon Univ. | Indoor photoreactor black light | 10 | Teflon (PTFE/FEP) | 288–313 | Stanier [89]; Robinson et al. [90] |
| Forschungazentrum Julich, Germany (SAPHIR) | Outdoor photoreactor | 270 | FEP | ambient | Rohrer et al. [68] |
| Forschungazentrum Julich, Germany | Dark chamber | 250 | PTFE/FEP | ambient | Mentel et al. [91]; Saathoff et al. [92] |
| Forschungazentrum Karlsruhe, Germany (AIDA) | Dark chamber (clouds) | 4–84 | Metal | 183–323 | Saathoff et al. [93]; Jonsson et al. [94] |
| Fundacion Centro de Estudios Ambientales del Mediterranean Spain (EUPHORE) | Outdoor photoreactor | 200 | FEP | ambient | Rodenas et al. [95]; Klotz et al. [96] |
| Leibniz Institute for Tropospheric Research Germany | Indoor photoreactor black light | 19 | FEP | 289–308 | Iinuma et al. [97] |
| Paul Scherrer Insitute Switzerland | Indoor photoreactor xenon arc | 27 | FEP | 290–298 | PSI [30]; Paulsen et al. [98] |
| University College Cork Ireland | Indoor photoreactor black light | 6.5 | FEP | 293–305 | Temime et al. [99]; Healy et al. [100] |
| University of Manchester UK | Indoor photoreactor Halogen/xenon arc | 18 | FEP | 288–313 | Centre for Atmos. Sci. [101] |
| University of California Riverside (CECERT) US | Indoor photoreacto xenon arc/black light | 90 (dual) | FEP | 278–323 | Carter et al. [85]; Song et al. [102] |
| University of California Riverside (APRC) US | Photoreactors (several indoor and outdoor) xenon arc/black light | 6–8 | PTFE/FEP | ambient | Dodge [36]; Tobias and Ziemann [103] |
| University of North Carolina US | Outdoor photoreactor | 120; 137 (dual); 150 (dual) | FEP | ambient | Jeffries [104,105,106]; Lee et al. [107] |
| US EPA | Indoor photoreactor Black light | 14.5 | FEP/TFE | 293-298 | Edney [108] |
| CSIRO | Outdoor photoreactor | 20 (dual) | FEP | ambient | Wiegand [109] |
| CSIRO Energy Technol Australia | Indoor photoreactor black light | 18 | FEP | ambient | Hynes et al. [110]; |
| CSIRO Australia | Indoor photoreactor black light | ~24 | FEP | ambient | White et al. [52] |
| CNRS-ICARE (HELIOS), France | Outdoor | 90 | FEP | ambient | Ren et al. [111] |
| Nat’l Inst. Environ. Studies Japan | Indoor photoreactor xenon arc | 6 | PFA | -- | Akimoto et al. [62] |
| Nat’l Inst. Environ. Studies Japan | Indoor photoreactor | 18 | FEP | ambient | Sato et al. [112] |
| Tsinghua University China | Indoor photoreactor | 2 | FEP | 283–333 | Wu et al. [113] |
| Key Lab. Organic Geochem. Guangzhou China | Indoor photoreactor black light | 30 | FEP | 283–313 | Wang et al., [34] |
| Key Lab. of Atmos. Inst. Comp. and Optical Radiation China | Indoor photoreactor | 0.83 | FEP | 298 | Hu et al. [114] |
| Kyungpook Natl Univ. Korea | Indoor photoreactor black light | 7 | FEP | 291–306 | Babar et al. [35] |
| Korea Institute of Science and Technol. | Indoor photoreactor black light | 5.8 (dual) | FEP | ambient | Bae et al. [31]; Moon et al. [115] |
| Functional Group | Structure | Change in Vapor Pressure (298 °K) a |
|---|---|---|
| Ketone | -C(O)- | 0.10 |
| Aldehyde | -C(O)H | 0.085 |
| Hydroxyl | -OH | 5.7 × 10−3 |
| Hydroperoxyl | -OOH | 2.5 × 10−3 |
| Nitrate | ONO2 | 6.8 × 10−3 |
| Carboxylic acid | -C(O)OH | 3.1 × 10−4 |
| Peroxyacid | -C(O)OOH | 3.2 × 10−3 |
| Acyl peroxynitrate | C(O)OONO2 | 2.7 × 10−3 |
| Extra carbon | -CH2- | 0.35 b |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hidy, G.M. Atmospheric Chemistry in a Box or a Bag. Atmosphere 2019, 10, 401. https://doi.org/10.3390/atmos10070401
Hidy GM. Atmospheric Chemistry in a Box or a Bag. Atmosphere. 2019; 10(7):401. https://doi.org/10.3390/atmos10070401
Chicago/Turabian StyleHidy, G. M. 2019. "Atmospheric Chemistry in a Box or a Bag" Atmosphere 10, no. 7: 401. https://doi.org/10.3390/atmos10070401
APA StyleHidy, G. M. (2019). Atmospheric Chemistry in a Box or a Bag. Atmosphere, 10(7), 401. https://doi.org/10.3390/atmos10070401