Comparison of Atmospheric Mercury Speciation at a Coastal and an Urban Site in Southeastern Texas, USA


Abstract
:1. Introduction
2. Experiments
2.1. Sampling Area
2.2. Measurements
3. Results and Discussion
3.1. Time Series of Atmospheric Mercury
3.2. Monthly Variations of Atmospheric Mercury
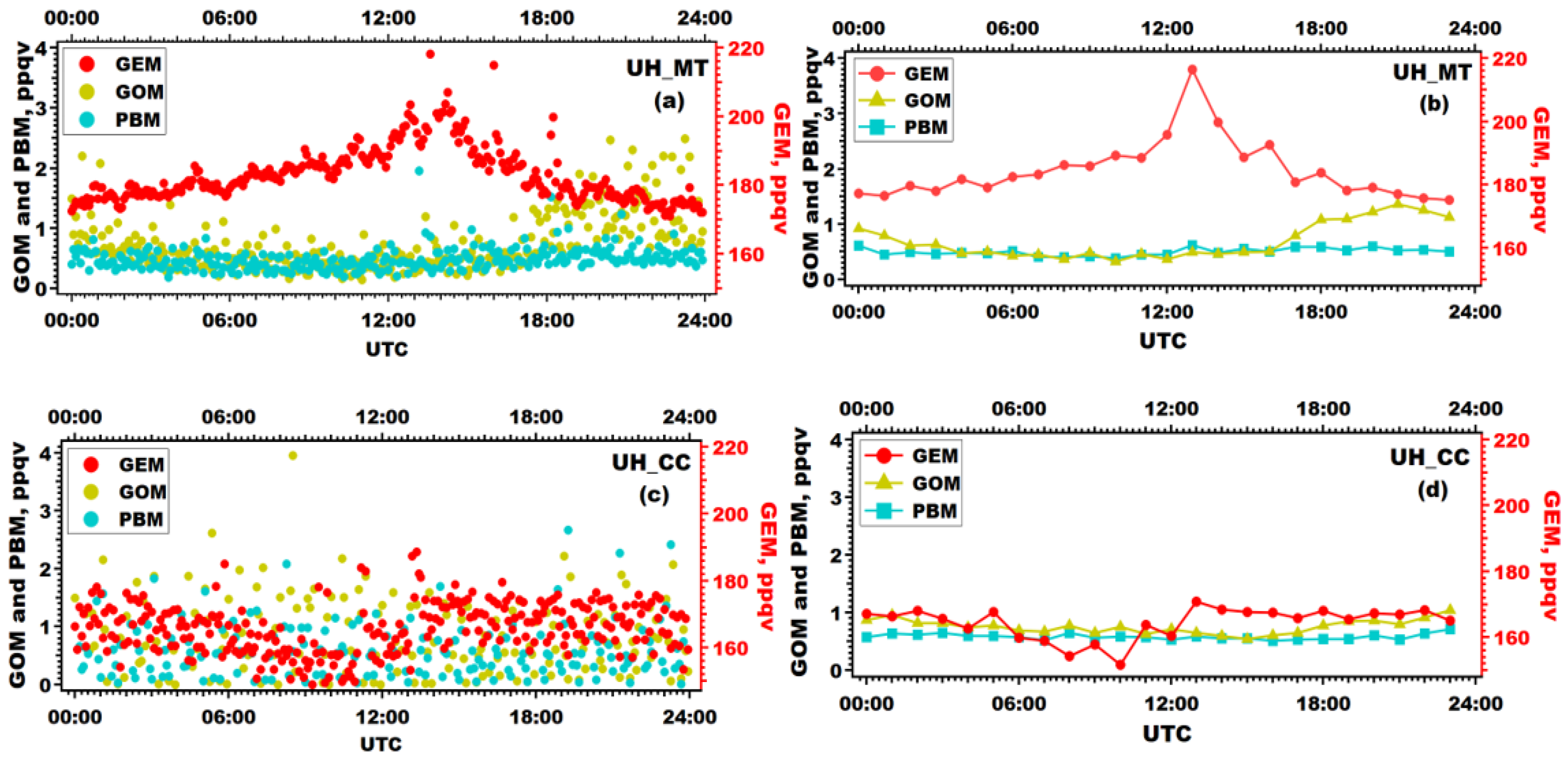
3.3. Seasonal and Diurnal Patterns of Atmospheric Mercury
3.4. Relationship between Atmospheric Mercury and Meteorological Parameters
Relationship between Atmospheric Mercury and Winds
3.5. Relationship with Key Trace Gases
3.6. Case Study of High Mercury Mixing Ratio Events
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pirrone, N.; Costa, P.; Pacyna, J.M.; Ferrara, R. Mercury emissions to the atmosphere from natural and anthropogenic sources in the Mediterranean region. Atmos. Environ. 2001, 35, 2997–3006. [Google Scholar] [CrossRef]
- Sprovieri, F.; Pirrone, N.; Ebinghaus, R.; Kock, H.; Dommergue, A. A review of worldwide atmospheric mercury measurements. Atmos. Chem. Phys. 2010, 10, 8245–8265. [Google Scholar] [CrossRef] [Green Version]
- United States Environmental Protection Agency. Health Effect of Mercury. Available online: http://www.epa.gov/hg/effects.html (accessed on 23 August 2019).
- Mao, H.; Talbot, R.; Sigler, J.M.; Sive, B.C.; Hegarty, J.D. Seasonal and diurnal variations of Hg over New England. Atmos. Chem. Phys. 2008, 33, L20808. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.D.; Jacob, D.J.; Yang, X. Global lifetime of elemental mercury against oxidation by atomic bromine in the free troposphere. Geophys. Res. Lett. 2006, 33, L20808. [Google Scholar] [CrossRef] [Green Version]
- Skov, H.; Brooks, S.B.; Goodsite, M.E.; Lindberg, S.E.; Meyers, T.P.; Landis, M.S.; Larsen, M.R.B.; Jensen, B.; McConville, G.; Christensen, J. Fluxes of reactive gaseous mercury measured with a newly developed method using relaxed eddy accumulation. Atmos. Environ. 2006, 40, 5452–5463. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 5, 809–822. [Google Scholar] [CrossRef]
- Gabriela, M.C.; Williamsona, D.G.; Brooks, S.; Lindberg, S. Atmospheric speciation of mercury in two contrasting Southeastern US airsheds. Atmos. Environ. 2005, 39, 4947–4958. [Google Scholar] [CrossRef]
- Laurier, F.J.; Mason, R.P.; Whalin, L.; Kato, S. Reactive gaseous mercury formation in the North Pacific Ocean’s marine boundary layer: A potential role of halogen chemistry. J. Geophys. Res. 2003. [Google Scholar] [CrossRef]
- Nair, U.S.; Wu, Y.; Walters, J.; Jansen, J.; Edgerton, E.S. Diurnal and seasonal variation of mercury species at coastal-suburban, urban, and rural sites in the southeastern United States. Atmos. Environ. 2011, 47, 499–508. [Google Scholar] [CrossRef]
- Duan, L.; Wang, X.; Wang, D.; Duan, Y.; Cheng, N.; Xiu, G. Atmospheric mercury speciation in Shanghai, China. Sci. Total Environ. 2017, 578, 460–468. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Jacob, D.J.; Horowitz, H.M.; Hu, L.; Sherwen, T.; Evans, M.J. Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury. J. Geophys. Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Valente, R.J.; Shea, C. Atmospheric mercury in the Great Smoky Mountains compared to regional and global levels. Atmos. Environ. 2007, 41, 1861–1873. [Google Scholar] [CrossRef]
- Lan, X.; Talbot, R.; Laine, P.; Lefer, B.; Flynn, J.; Sun, Z.; Torres, A. Seasonal and diurnal variations of total gaseous mercury in urban Houston, Texas (USA). Atmosphere 2014, 5, 399–419. [Google Scholar] [CrossRef] [Green Version]
- Cheng, I.; Zhang, L.; Mao, H.; Blanchard, P.; Tordon, R.; Dalziel, J. Seasonal and diurnal patterns of speciated atmospheric mercury at a coastal-rural and a coastal-urban site. Atmos. Environ. 2014, 82, 193–205. [Google Scholar] [CrossRef]
- Rutter, A.P.; Snyder, D.C.; Stone, E.A.; Schauer, J.J. In situ measurements of speciated atmospheric mercury and the identification of source regions in the Mexico City Metropolitan Area. Atmos. Chem. Phys. 2007, 9, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Talbot, R. Speciated mercury at marine, coastal, and inland sites in New England—Part 1: Temporal variability. Atmos. Chem. Phys. 2012, 12, 5099–5112. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Talbot, R.; Castro, M.; Perry, K.; Luke, W. Seasonal and diurnal variations of atmospheric mercury across the US determined from AMNet monitoring data. Atmos. Chem. Phys. 2012, 12, 10569–10582. [Google Scholar] [CrossRef] [Green Version]
- Engle, M.A.; Tate, M.T.; Krabbenhoft, D.P.; Schauer, J.J.; Kolker, A.; Shanley, J.B.; Bothner, M.H. Comparison of atmospheric mercury speciation and deposition at nine sites across central and eastern North America. J. Geophys. Res. 2010, 115, D18306. [Google Scholar] [CrossRef]
- Malcolm, E.G.; Keeler, G.J.; Landis, M.S. The effects of the coastal environment on the atmospheric mercury cycle. J. Geophys. Res. 2003. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Talbot, R.; Lan, X. Influence of Climate Change and Meteorological Factors on Houston’s Air Pollution: Ozone a Case Study. Atmosphere 2015, 6, 623–640. [Google Scholar] [CrossRef] [Green Version]
- Banta, R.M.; Senff, C.J.; Gammon, J.N.; Darby, L.S.; Ryerson, T.B.; Alvarez, R.J.; Sandberg, S.P.; Williams, E.J.; Trainer, M. A bad air day in Houston. Bull. Am. Meteorol. Soc. 2005, 86, 657–669. [Google Scholar] [CrossRef]
- Brooks, S.; Luke, W.; Cohen, M.; Kelly, P.; Lefer, B.; Rappengluck, B. Mercury species measured atop the Moody Tower TRAMP site, Houston, Texas. Atmos. Environ. 2010, 44, 4045–4055. [Google Scholar] [CrossRef]
- Ren, X.; Luke, W.; Kelley, P.; Cohen, M.; Ngan, F.; Artz, R.; Walker, J.; Brooks, S.; Moore, C.; Swartzendruber, P.; et al. Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011. Atmosphere 2014, 5, 230–251. [Google Scholar] [CrossRef] [Green Version]
- Sigler, J.M.; Mao, H.; Sive, B.C.; Talbot, R. Oceanic influence on atmospheric mercury at coastal and inland sites: A springtime noreaster in New England. Atmos. Chem. Phys. 2009, 9, 4023–4030. [Google Scholar] [CrossRef] [Green Version]
- Gratz, L.; Keeler, G.; Marsik, F.; Barres, J.; Dvonch, J. Atmospheric transport of speciated mercury across southern Lake Michigan: Influence from emission sources in the Chicago/Gary urban area. Sci. Total Environ. 2013, 448, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Obrist, D.; Tas, E.; Peleg, M.; Matveev, V.; Faïn, X.; Asaf, D.; Luria, M. Bromine-induced oxidation of mercury in the mid-latitude atmosphere. Nat. Geosci. 2011, 4, 22–26. [Google Scholar] [CrossRef]
- Liu, B.; Keeler, G.J.; Dvonch, J.T.; Barres, J.A.; Lynam, M.M.; Marsik, F.J.; Morgan, T.J. Temporal variability of mercury speciation in urban air. Atmos. Environ. 2007, 10, 063. [Google Scholar] [CrossRef]
- Zhu, J.L.; Wang, T.; Talbot, R.; Mao, H. Characteristics of atmospheric Total Gaseous Mercury (TGM) observed in urban Nanjing, China. Atmos. Chem. Phys. 2012, 12, 12103–12118. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, H.; Wang, D. Spatial and temporal distribution of gaseous elemental mercury in Chongqing, China. Environ. Monit. Assess. 2009, 156, 479–489. [Google Scholar] [CrossRef]
- Liu, B.; Keeler, G.; Timothy Dvonch, J.; Barres, J.; Lynam, M.; Marsik, F.; Morgan, J. Urban–rural differences in atmospheric mercury speciation. Atmos. Environ. 2010, 44, 2013–2023. [Google Scholar] [CrossRef]
- Lefer, B.; Rappenglück, B.; Flynn, J.; Haman, C. Photochemical and meteorological relationships during the Texas-II Radical and Aerosol Measurement Project (TRAMP). Atmos. Environ. 2010, 44, 4005–4013. [Google Scholar] [CrossRef]
- Ryerson, T.B.; Trainer, M.; Angevine, W.M.; Brock, C.A.; Dissly, R.W.; Fehsenfeld, F.C.; Frost, G.J.; Goldan, P.D.; Holloway, J.S.; Hubler, G.; et al. Effect of petrochemical industrial emissions of reactive alkenes and NOx on tropospheric ozone formation in Houston, Texas. J. Geophys. Res. 2003, 108, 4249. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Moody Tower (UH_MT) | Coastal Center (UH_CC) | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean ± Std | Median | Range | N | Mean ± Std | Median | Range | N | |
| GEM (ppqv) | ||||||||
| 2012 spring | 182 ± 17 | 180 | 81~973 | 7796 | * | * | * | * |
| 2012 summer | 189 ± 15 | 179 | 94~1100 | 11,483 | 179 ± 36 | 176 | 11~498 | 5137 |
| 2012 fall | 183 ± 26 | 172 | 113~27,300 | 10,690 | 197 ± 29 | 173 | 11~2230 | 12,559 |
| 2012 winter | 182 ± 18 | 179 | 112~2690 | 11,405 | 174 ± 24 | 170 | 12~754 | 10,164 |
| 2013 spring | 184 ± 23 | 180 | 96~4630 | 10,348 | 113 ± 19 | 124 | 12~2820 | 11,297 |
| Total | 184 ± 30 | 178 | 81~27,300 | 51,723 | 164 ± 34 | 162 | 11~2820 | 39,160 |
| GOM (ppqv) | ||||||||
| 2012 spring | 1.09 ± 0.5 | 0.64 | 0.10~12.39 | 590 | * | * | * | * |
| 2012 summer | 0.88 ± 0.3 | 0.42 | 0.18~26.04 | 874 | 0.46 ± 0.4 | 0.26 | 0.15~3.15 | 407 |
| 2012 fall | 0.66 ± 0.1 | 0.47 | 0.21~9.23 | 944 | 0.69 ± 1.1 | 0.18 | 0.1~10.71 | 1042 |
| 2012 winter | 0.51 ± 1.6 | 0.29 | 0.19~11.11 | 811 | 0.66 ± 0.7 | 0.03 | 0.1~8.52 | 842 |
| 2013 spring | 0.48 ± 1.2 | 0.17 | 0.14~11.57 | 756 | 1.09 ± 2.1 | 0.89 | 0.1~10.76 | 790 |
| Total | 0.72 ± 0.9 | 0.39 | 0.10~26.04 | 3856 | 0.75 ± 0.3 | 0.23 | 0.1~10.76 | 3082 |
| PBM (ppqv) | ||||||||
| 2012 spring | 0.47 ± 1.9 | 0.41 | 0.18~6.01 | 661 | * | * | * | * |
| 2012 summer | 0.45 ± 1.3 | 0.29 | 0.11~5.53 | 880 | 0.19 ± 0.2 | 0.12 | 0.12~4.69 | 407 |
| 2012 fall | 0.47 ± 1.6 | 0.36 | 0.15~6.73 | 955 | 0.59 ± 0.9 | 0.26 | 0.14~7.55 | 1042 |
| 2012 winter | 0.57 ± 0.8 | 0.41 | 0.21~4.86 | 836 | 0.67 ± 1.1 | 0.21 | 0.11~7.27 | 842 |
| 2013 spring | 0.57 ± 1.0 | 0.31 | 0.17~17.65 | 523 | 0.67 ± 0.8 | 0.55 | 0.16~5.96 | 790 |
| Total | 0.71 ± 0.1 | 0.38 | 0.10~17.65 | 3976 | 0.58 ± 1.6 | 0.24 | 0.10~7.55 | 3082 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griggs, T.; Liu, L.; Talbot, R.W.; Torres, A.; Lan, X. Comparison of Atmospheric Mercury Speciation at a Coastal and an Urban Site in Southeastern Texas, USA. Atmosphere 2020, 11, 73. https://doi.org/10.3390/atmos11010073
Griggs T, Liu L, Talbot RW, Torres A, Lan X. Comparison of Atmospheric Mercury Speciation at a Coastal and an Urban Site in Southeastern Texas, USA. Atmosphere. 2020; 11(1):73. https://doi.org/10.3390/atmos11010073
Chicago/Turabian StyleGriggs, Travis, Lei Liu, Robert W. Talbot, Azucena Torres, and Xin Lan. 2020. "Comparison of Atmospheric Mercury Speciation at a Coastal and an Urban Site in Southeastern Texas, USA" Atmosphere 11, no. 1: 73. https://doi.org/10.3390/atmos11010073