Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece


Abstract
:1. Introduction
2. Experiments
2.1. Experimental Design
2.2. PMF Analysis
2.3. Trajectory Analysis and Long-Range Transport
3. Results
3.1. PM2.5 Chemical Composition
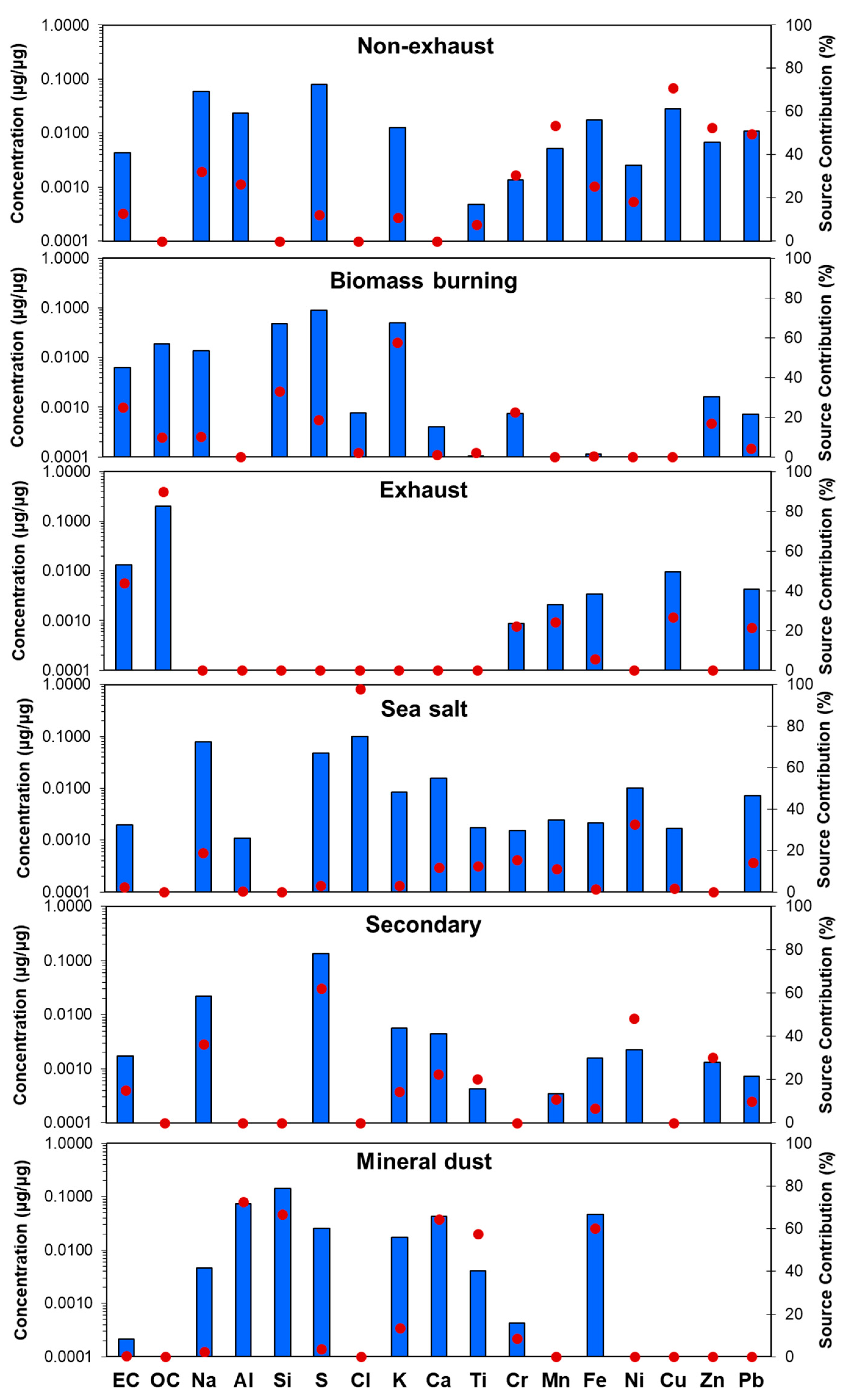
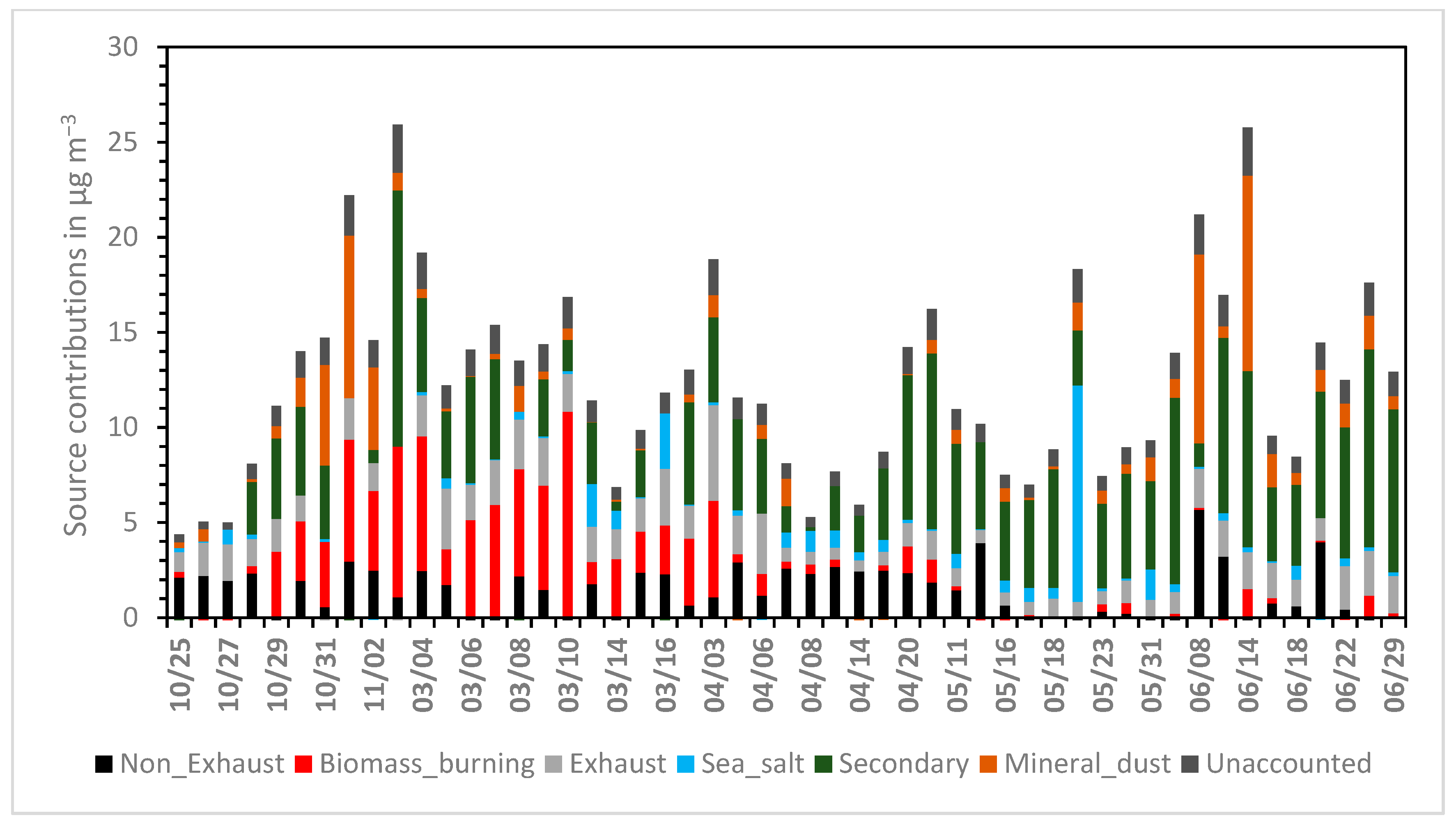
3.2. PM2.5 Source Apportionment
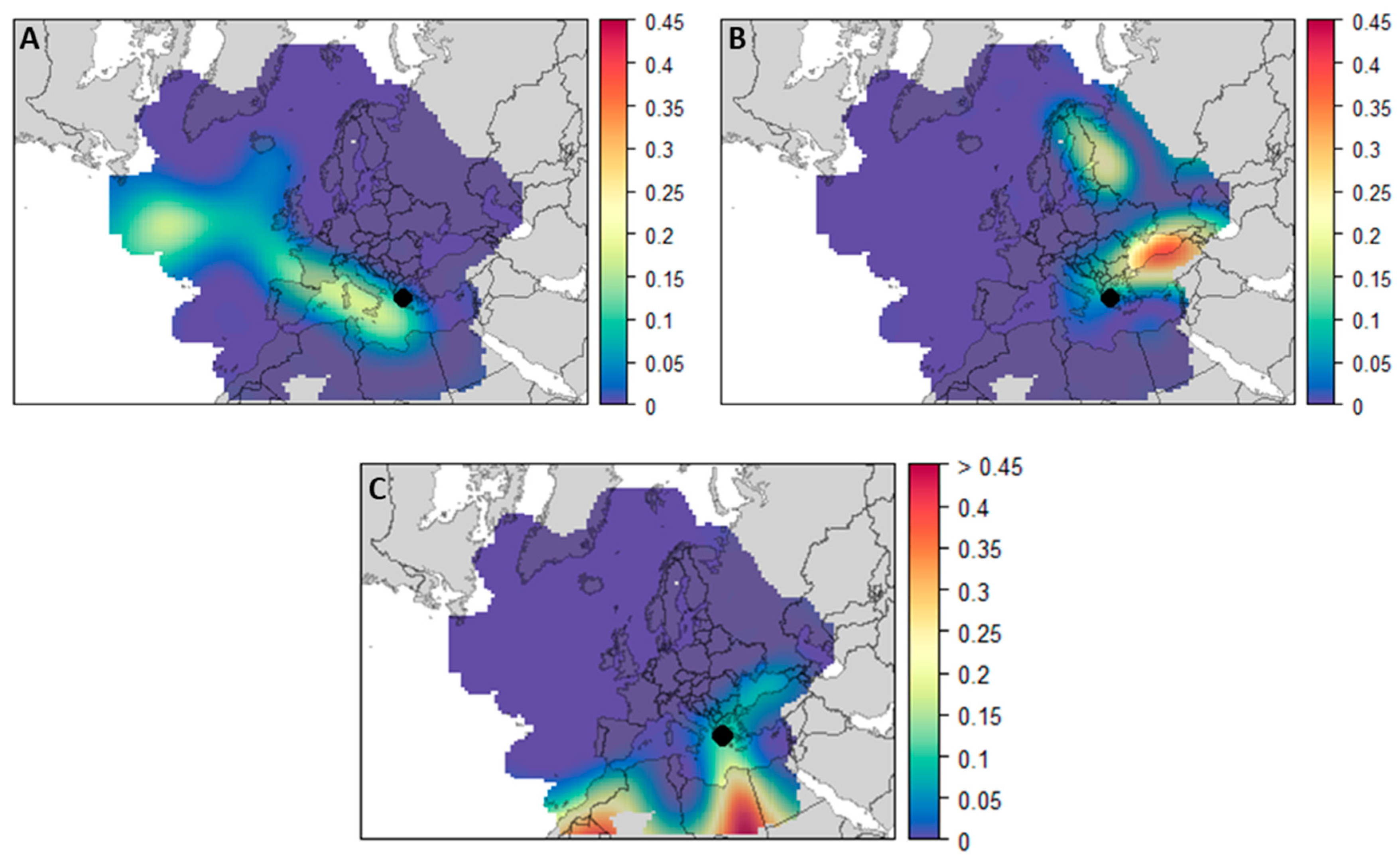
3.3. The Role of Long-Range Transport
3.4. Nonrefractory PM1 Chemical Composition
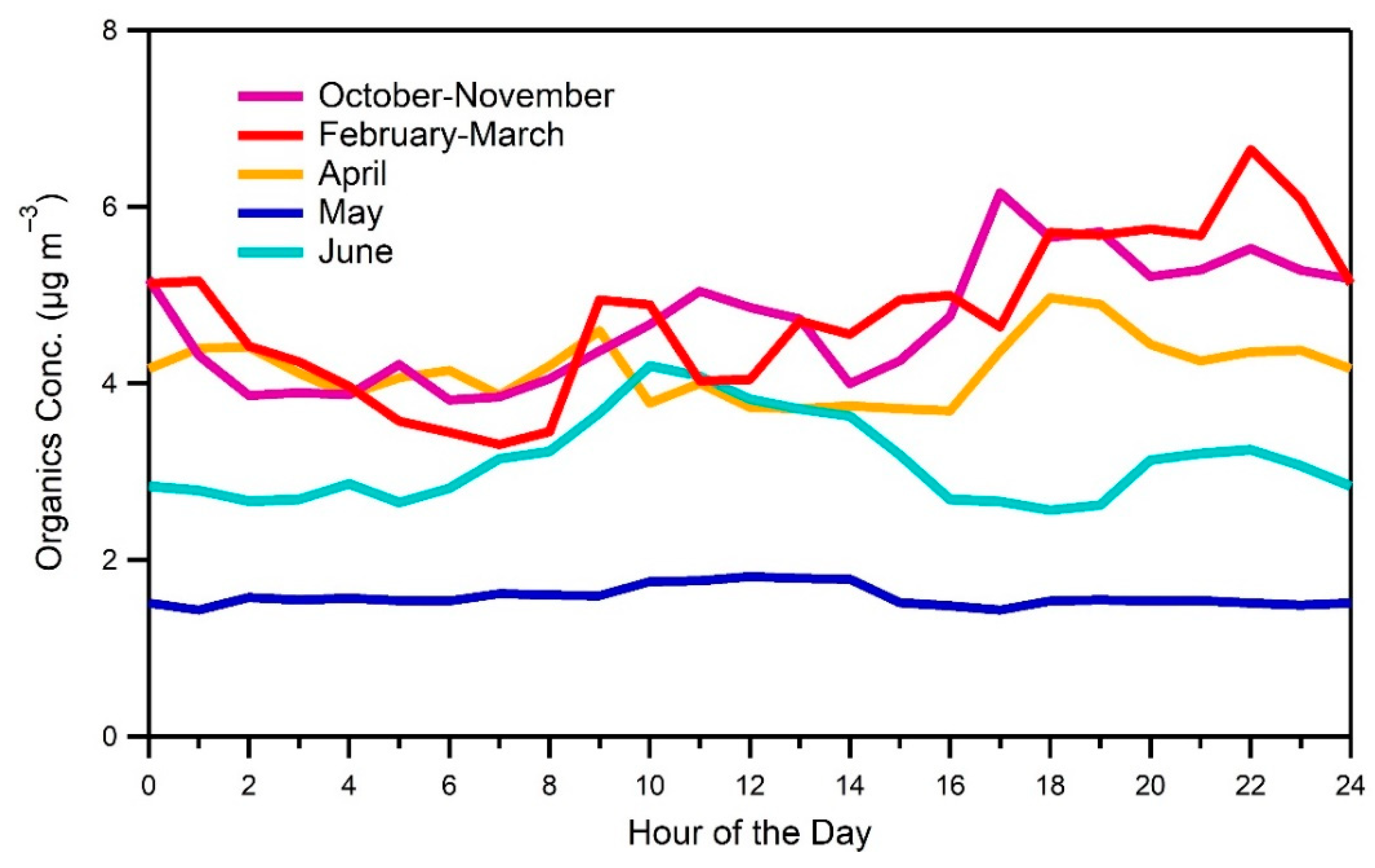
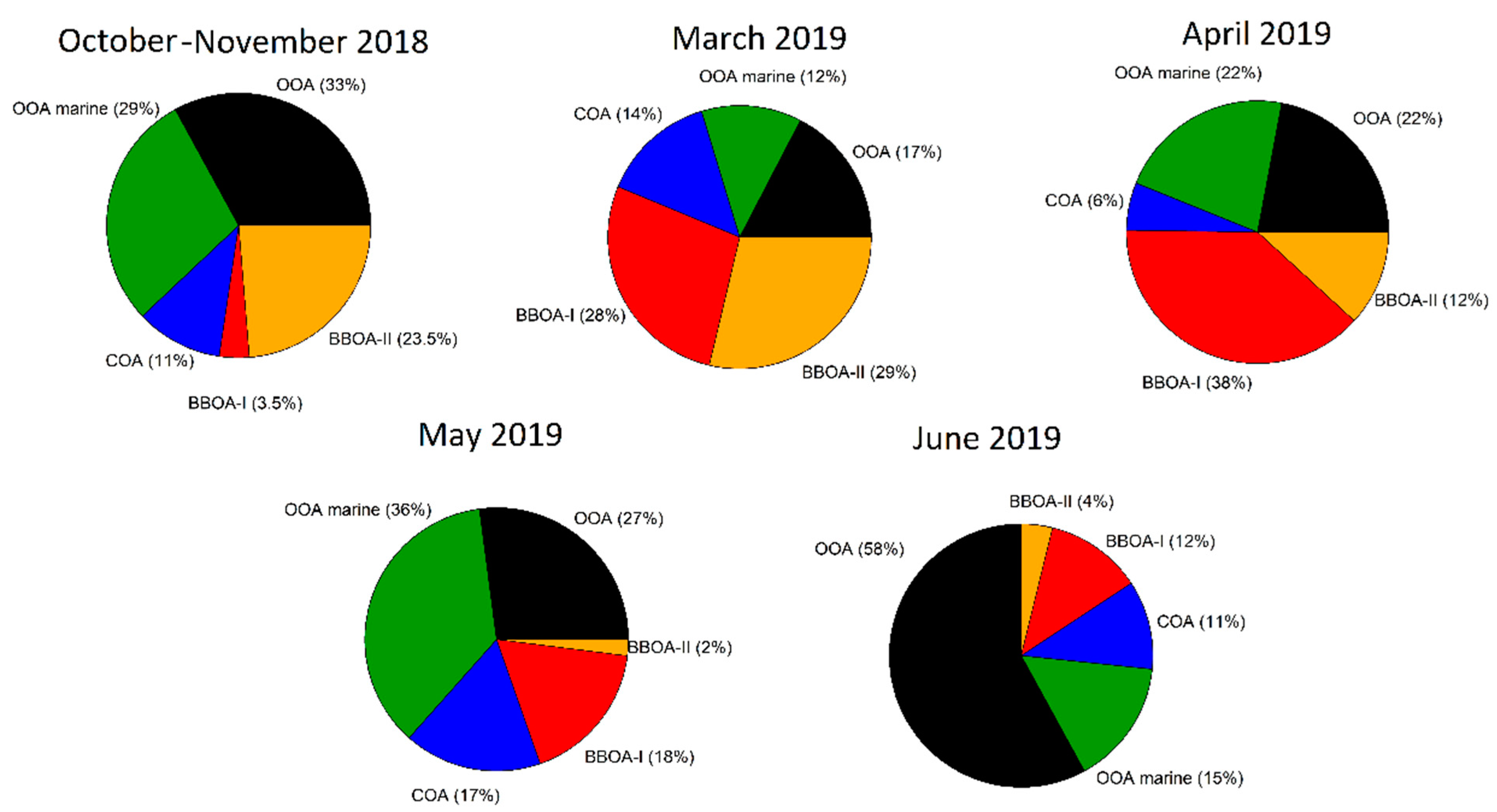
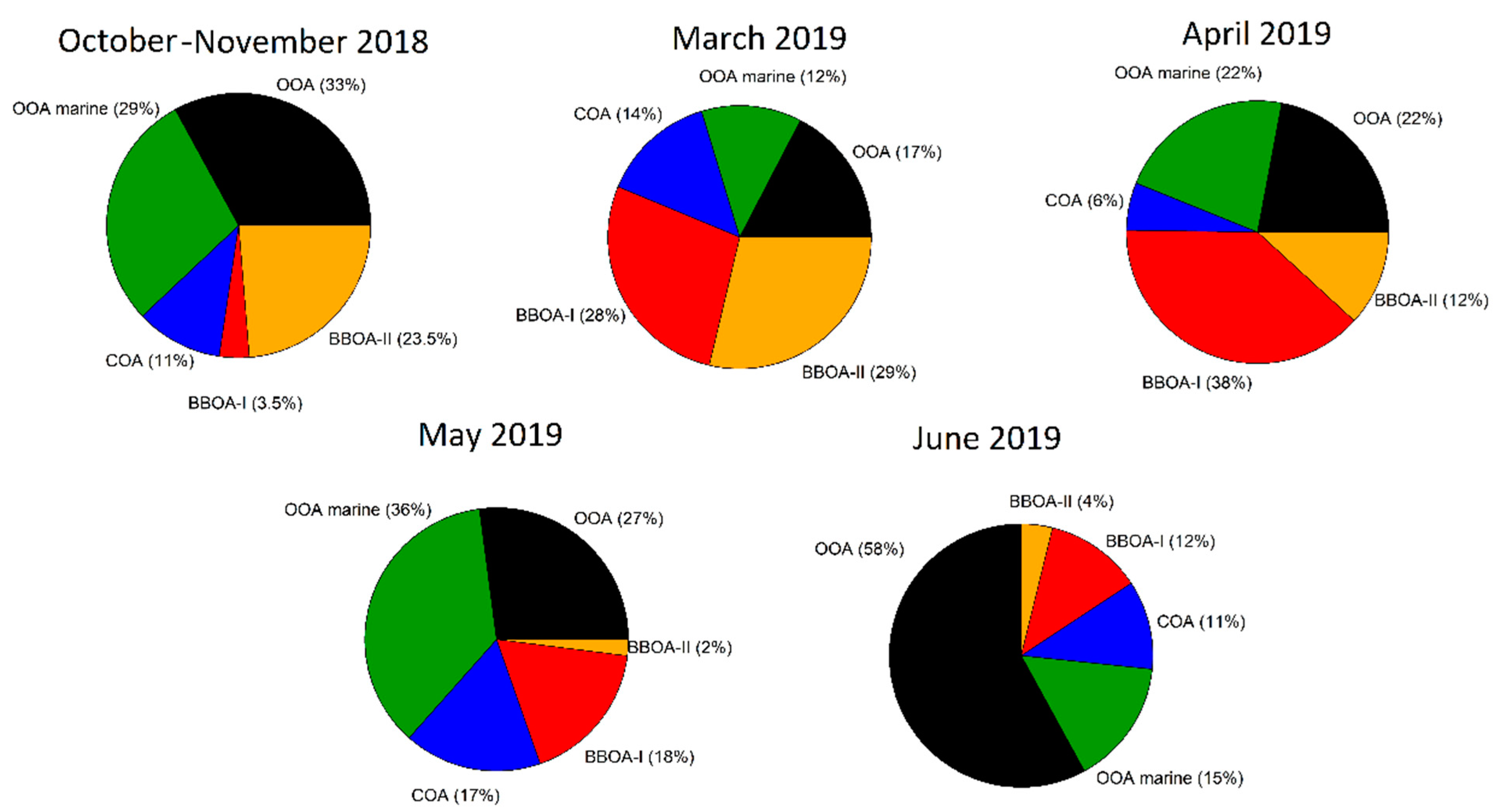
3.5. Sources of OA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Samoli, E.; Stafoggia, M.; Rodopoulou, S.; Ostro, B.; Declercq, C.; Alessandrini, E.; Díaz, J.; Karanasiou, A.; Kelessis, A.G.; Le Tertre, A.; et al. Associations between fine and coarse particles and mortality in Mediterranean cities: Results from the MED-PARTICLES Project. Environ. Health Perspect. 2013, 121, 932–938. [Google Scholar] [CrossRef]
- Samoli, E.; Stafoggia, M.; Rodopoulou, S.; Ostro, B.; Alessandrini, E.; Basagaña, X.; Díaz, J.; Faustini, A.; Gandini, M.; Karanasiou, A.; et al. Which specific causes of death are associated with short term exposure to fine and coarse particles in Southern Europe? Results from the MED-PARTICLES project. Environ. Int. 2014, 67, 54–61. [Google Scholar] [CrossRef]
- Katsouyanni, K.; Touloumi, G.; Samoli, E.; Gryparis, A.; Le Tertre, A.; Monopolis, Y.; Rossi, G.; Zmirou, D.; Ballester, F.; Boumghar, A.; et al. Confounding and effect modification in the short-term effects of ambient particles on total mortality: Results from 29 European cities within the APHEA2 Project. Epidemiology 2001, 12, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Taghvaee, S.; Sowlat, M.H.; Diapouli, E.; Manousakas, M.I.; Vasilatou, V.; Eleftheriadis, K.; Sioutas, C. Source apportionment of the oxidative potential of fine ambient particulate matter (PM2.5) in Athens, Greece. Sci. Total Environ. 2019, 653, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Cesari, D.; Donateo, A.; Conte, M.; Contini, D. Inter-comparison of source apportionment of PM10 using PMF and CMB in three sites nearby an industrial area in central Italy. Atmos. Res. 2016, 182, 282–293. [Google Scholar] [CrossRef]
- Belis, C.A.; Karagulian, F.; Larsen, B.R.; Hopke, P.K. Critical review and meta-analysis of ambient particulate matter source apportionment using receptor models in Europe. Atmos. Environ. 2013, 69, 94–108. [Google Scholar] [CrossRef]
- Amato, F.; Alastuey, A.; Karanasiou, A.; Lucarelli, F.; Nava, S.; Calzolai, G.; Severi, M.; Becagli, S.; Gianelle, V.L.; Colombi, C.; et al. AIRUSE-LIFE +: A harmonized PM speciation and source apportionment in five southern European cities. Atmos. Chem. Phys. 2016, 16, 3289–3309. [Google Scholar] [CrossRef] [Green Version]
- Manousakas, M.; Diapouli, E.; Papaefthymiou, H.; Kantarelou, V.; Zarkadas, C.; Kalogridis, A.-C.; Karydas, A.G.; Eleftheriadis, K. XRF characterization and source apportionment of PM10 samples collected in a coastal city. X-ray Spectrom. 2018, 47, 190–200. [Google Scholar] [CrossRef]
- Belis, C.A.; Pernigotti, D.; Pirovano, G.; Favez, O.; Jaffrezo, J.L.; Kuenen, J.; Denier van Der Gon, H.; Reizer, M.; Riffault, V.; Alleman, L.Y.; et al. Evaluation of receptor and chemical transport models for PM10 source apportionment. Atmos. Environ. X 2020, 5, 100053. [Google Scholar] [CrossRef]
- Gunchin, G.; Manousakas, M.; Osan, J.; Karydas, A.G.; Eleftheriadis, K.; Lodoysamba, S.; Shagjjamba, D.; Migliori, A.; Padilla-Alvarez, R.; Streli, C.; et al. Three-year long source apportionment study of airborne particles in Ulaanbaatar using X-ray fluorescence and Positive Matrix Factorization. Aerosol Air Qual. Res. 2019, 5, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Favez, O.; El Haddad, I.; Piot, C.; Borave, A.; Abidi, E.; Marchand, N.; Jaffrezo, J.L.; Besombes, J.L.; Personnaz, M.B.; Sciare, J.; et al. Inter-comparison of source apportionment models for the estimation of wood burning aerosols during wintertime in an Alpine city (Grenoble, France). Atmos. Chem. Phys. 2010, 10, 5295–5314. [Google Scholar] [CrossRef] [Green Version]
- Paatero, P.; Tappert, U. Positive Matrix Factorization: A non-negative factor model with optimal utilization of error stimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Crippa, M.; Canonaco, F.; Lanz, V.A.; Äijälä, M.; Allan, J.D.; Carbone, S.; Capes, G.; Ceburnis, D.; Dall’Osto, M.; Day, D.A.; et al. Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach. Atmos. Chem. Phys. 2014, 14, 6159–6176. [Google Scholar] [CrossRef] [Green Version]
- Petit, J.E.; Favez, O.; Sciare, J.; Canonaco, F.; Croteau, P.; Močnik, G.; Jayne, J.; Worsnop, D.; Leoz-Garziandia, E. Submicron aerosol source apportionment of wintertime pollution in Paris, France by double positive matrix factorization (PMF2) using an aerosol chemical speciation monitor (ACSM) and a multi-wavelength Aethalometer. Atmos. Chem. Phys. 2014, 14, 13773–13787. [Google Scholar] [CrossRef] [Green Version]
- Manousakas, M.; Diapouli, E.; Papaefthymiou, H.; Migliori, A.; Karydas, A.G.; Padilla-Alvarez, R.; Bogovac, M.; Kaiser, R.B.; Jaksic, M.; Bogdanovic-Radovic, I.; et al. Source apportionment by PMF on elemental concentrations obtained by PIXE analysis of PM10 samples collected at the vicinity of lignite power plants and mines in Megalopolis, Greece. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2015, 349, 114–124. [Google Scholar] [CrossRef]
- Karanasiou, A.; Querol, X.; Alastuey, A.; Perez, N.; Pey, J.; Perrino, C.; Berti, G.; Gandini, M.; Poluzzi, V.; Ferrari, S.; et al. Particulate matter and gaseous pollutants in the Mediterranean Basin: Results from the MED-PARTICLES project. Sci. Total Environ. 2014, 488, 297–315. [Google Scholar] [CrossRef]
- Karanasiou, A.; Mihalopoulos, N. Air quality in urban Environments in the Eastern Mediterranean. In The Handbook of Environmental Chemistry 26; Viana, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 219–238. [Google Scholar]
- Tsiflikiotou, M.A.; Kostenidou, E.; Papanastasiou, D.K.; Patoulias, D.; Zarmpas, P.; Paraskevopoulou, D.; Diapouli, E.; Kaltsonoudis, C.; Florou, K.; Bougiatioti, A.; et al. Summertime particulate matter and its composition in Greece. Atmos. Environ. 2019, 213, 597–607. [Google Scholar] [CrossRef]
- Kostenidou, E.; Florou, K.; Kaltsonoudis, C.; Tsiflikiotou, M.; Vratolis, S.; Eleftheriadis, K.; Pandis, S.N. Sources and chemical characterization of organic aerosol during the summer in the eastern Mediterranean. Atmos. Chem. Phys. 2015, 15, 11355–11371. [Google Scholar] [CrossRef] [Green Version]
- Kostenidou, E.; Kaltsonoudis, C.; Tsiflikiotou, M.; Louvaris, E.; Russell, L.M.; Pandis, S.N. Burning of olive tree branches: A major organic aerosol source in the Mediterranean. Atmos. Chem. Phys. Discuss. 2013, 13, 7223–7266. [Google Scholar] [CrossRef]
- Pikridas, M.; Tasoglou, A.; Florou, K.; Pandis, S.N. Characterization of the origin of fine particulate matter in a medium size urban area in the Mediterranean. Atmos. Environ. 2013, 80, 264–274. [Google Scholar] [CrossRef]
- Florou, K.; Papanastasiou, D.K.; Pikridas, M.; Kaltsonoudis, C.; Louvaris, E.; Gkatzelis, G.I.; Patoulias, D.; Mihalopoulos, N.; Pandis, S.N. The contribution of wood burning and other pollution sources to wintertime organic aerosol levels in two Greek cities. Atmos. Chem. Phys. 2017, 17, 3145–3163. [Google Scholar] [CrossRef] [Green Version]
- Saffari, A.; Daher, N.; Samara, C.; Voutsa, D.; Kouras, A.; Manoli, E.; Karagkiozidou, O.; Vlachokostas, C.; Moussiopoulos, N.; Shafer, M.M.; et al. Increased biomass burning due to the economic crisis in Greece and its adverse impact on wintertime air quality in Thessaloniki. Environ. Sci. Technol. 2013, 47, 13313–13320. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef] [Green Version]
- Diapouli, E.; Manousakas, M.; Vratolis, S.; Vasilatou, V.; Maggos, T.; Saraga, D.; Grigoratos, T.; Argyropoulos, G.; Voutsa, D.; Samara, C.; et al. Evolution of air pollution source contributions over one decade, derived by PM 10 and PM 2.5 source apportionment in two metropolitan urban areas in Greece. Atmos. Environ. 2017, 164, 416–430. [Google Scholar] [CrossRef]
- Cavalli, F.; Viana, M.; Yttri, K.E.; Genberg, J.; Putaud, J.-P. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: The EUSAAR protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Carslaw, D.C.; Ropkins, K. Openair—An R package for air quality data analysis. Environ. Model. Softw. 2012, 27, 52–61. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. Noaa’s hysplit atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time environmental applications and display system: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Stohl, A. Trajectory statistics—A new method to establish source-receptor relationships of air pollutants and its application to the transport of particulate sulfate in Europe. Atmos. Environ. 1996, 30, 579–587. [Google Scholar] [CrossRef]
- Remoundaki, E.; Kassomenos, P.; Mantas, E.; Mihalopoulos, N.; Tsezos, M. Composition and mass closure of PM2.5 in urban environment (Athens, Greece). Aerosol Air Qual. Res. 2013, 13, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Samara, C.; Voutsa, D.; Kouras, A.; Eleftheriadis, K.; Maggos, T.; Saraga, D.; Petrakakis, M. Organic and elemental carbon associated to PM10 and PM 2.5 at urban sites of northern Greece. Environ. Sci. Pollut. Res. Int. 2014, 21, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.; Faria, T.; Diapouli, E.; Manousakas, M.I.; Eleftheriadis, K.; Viana, M.; Almeida, S.M. Relationship between indoor and outdoor size-fractionated particulate matter in urban microenvironments: Levels, chemical composition and sources. Environ. Res. 2020, 183, 109203. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Chi, X.; Maenhaut, W.; Querol, X.; Alastuey, A.; Mikuska, P.; Vecera, Z. Organic and elemental carbon concentrations in carbonaceous aerosols during summer and winter sampling campaigns in Barcelona, Spain. Atmos. Environ. 2006, 40, 2180–2193. [Google Scholar] [CrossRef]
- Belis, C.A.; Larsen, B.R.; Amato, F.; Haddad, I.E.; Favez, O.; Harrison, R.M.; Hopke, P.K.; Nava, S.; Paatero, P.; Prévôt, A.; et al. Air Pollution Source Apportionment with Receptor Models; Europena Union: Ispra, Italy, 2014. [Google Scholar]
- Manousakas, M.; Papaefthymiou, H.; Diapouli, E.; Migliori, A.; Karydas, A.G.; Bogdanovic-Radovic, I.; Eleftheriadis, K. Assessment of PM2.5 sources and their corresponding level of uncertainty in a coastal urban area using EPA PMF 5.0 enhanced diagnostics. Sci. Total Environ. 2017, 574, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pateraki, S.; Manousakas, M.; Bairachtari, K.; Kantarelou, V.; Eleftheriadis, K.; Vasilakos, C.; Assimakopoulos, V.D.; Maggos, T. The traffic signature on the vertical PM profile: Environmental and health risks within an urban roadside environment. Sci. Total Environ. 2019, 646, 448–459. [Google Scholar] [CrossRef]
- Kim, E.; Hopke, P.K. Source characterization of ambient fine particles at multiple sites in the Seattle area. Atmos. Environ. 2008, 42, 6047–6056. [Google Scholar] [CrossRef]
- Vasilatou, V.; Manousakas, M.; Gini, M.; Diapouli, E.; Scoullos, M.; Eleftheriadis, K. Long term flux of Saharan dust to the Aegean sea around the Attica region, Greece. Front. Mar. Sci. 2017, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Say, N.P. Lignite-fired thermal power plants and SO2 pollution in Turkey. Energy Policy 2006, 34, 2690–2701. [Google Scholar] [CrossRef]
- Mitsakou, C.; Kallos, G.; Papantoniou, N.; Spyrou, C.; Solomos, S.; Astitha, M.; Housiadas, C. Saharan dust levels in Greece and received inhalation doses. Atmos. Chem. Phys. Discuss. 2008, 8, 11967–11996. [Google Scholar] [CrossRef] [Green Version]
- Nava, S.; Becagli, S.; Calzolai, G.; Chiari, M.; Lucarelli, F.; Prati, P.; Traversi, R.; Udisti, R.; Valli, G.; Vecchi, R. Saharan dust impact in central Italy: An overview on three years elemental data records. Atmos. Environ. 2012, 60, 444–452. [Google Scholar] [CrossRef]
- Rodriguez, S.; Querol, X.; Alastuey, A.; Kallos, G.; Kakaliagou, O. Saharan dust contributions to PM10 and TSP levels in Southern and Eastern Spain. Atmos. Environ. 2001, 35, 2433–2447. [Google Scholar] [CrossRef]
- Eleftheriadis, K.; Ochsenkuhn, K.M.; Lymperopoulou, T.; Karanasiou, A.; Razos, P.; Ochsenkuhn-Petropoulou, M. Influence of local and regional sources on the observed spatial and temporal variability of size resolved atmospheric aerosol mass concentrations and water-soluble species in the Athens metropolitan area. Atmos. Environ. 2014, 97, 252–261. [Google Scholar] [CrossRef]
- Kuwata, M.; Zorn, S.R.; Martin, S.T. Using elemental ratios to predict the density of organic material composed of carbon, hydrogen, and oxygen. Environ. Sci. Technol. 2012, 46, 787–794. [Google Scholar] [CrossRef]
- Huang, S.; Wu, Z.; Poulain, L.; Van Pinxteren, M.; Merkel, M.; Assmann, D.; Herrmann, H.; Wiedensohler, A. Source apportionment of the organic aerosol over the Atlantic Ocean from 53° N to 53° S: Significant contributions from marine emissions and long-range transport. Atmos. Chem. Phys. 2018, 18, 18043–18062. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PM2.5 | EC | OC | Na | Mg | Al | Si | S | Cl | K | Ca | |
| Average | 12.40 | 0.38 | 2.85 | 0.35 | <0.10 | 0.29 | 0.71 | 0.92 | 0.12 | 0.17 | 0.10 |
| Standard Deviation | 6.46 | 0.20 | 1.63 | 0.27 | - | 0.29 | 0.64 | 0.47 | 0.31 | 0.14 | 0.13 |
| Ti | V | Cr | Mn | Fe | Ni | Cu | Zn | Br | Sr | Pb | |
| Average | 0.02 | <0.01 | 0.01 | 0.02 | 0.10 | 0.02 | 0.06 | 0.05 | <0.05 | <0.05 | 0.04 |
| Standard Deviation | 0.02 | - | 0.00 | 0.00 | 0.11 | 0.01 | 0.03 | 0.05 | - | - | 0.01 |
| Ammonium (μg m−3) | Sulfate (μg m−3) | Organics (μg m−3) | Nitrate (μg m−3) | Chloride (μg m−3) | NR-PM1 (μg m−3) | |
|---|---|---|---|---|---|---|
| October–November 2018 | 1.2 | 3.7 | 4.7 | 0.24 | 0.05 | 9.9 |
| February–March 2018 | 0.95 | 2.5 | 4.7 | 0.45 | 0.09 | 8.7 |
| April 2019 | 1.04 | 2.8 | 4.2 | 0.22 | 0.03 | 8.3 |
| May 2019 | 0.6 | 1.6 | 1.6 | 0.09 | 0.02 | 3.8 |
| June 2019 | 0.96 | 2.7 | 3.1 | 0.1 | 0.01 | 6.9 |
| Average | 0.96 | 2.7 | 3.7 | 0.23 | 0.04 | 7.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manousakas, M.I.; Florou, K.; Pandis, S.N. Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece. Atmosphere 2020, 11, 330. https://doi.org/10.3390/atmos11040330
Manousakas MI, Florou K, Pandis SN. Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece. Atmosphere. 2020; 11(4):330. https://doi.org/10.3390/atmos11040330
Chicago/Turabian StyleManousakas, Manousos Ioannis, Kalliopi Florou, and Spyros N. Pandis. 2020. "Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece" Atmosphere 11, no. 4: 330. https://doi.org/10.3390/atmos11040330