Gas-Phase Reaction of trans-2-Methyl-2-butenal with Cl: Kinetics, Gaseous Products, and SOA Formation


Abstract
:1. Introduction
2. Experiments
2.1. Kinetic Study
2.2. Gaseous Products Study
2.3. SOA Formation Study
2.4. Chemicals
3. Results
3.1. Determination of the Rate Coefficient
3.2. Identification of the Reaction Products in the Gas-Phase
3.2.1. Detection by SPME/GC-MS
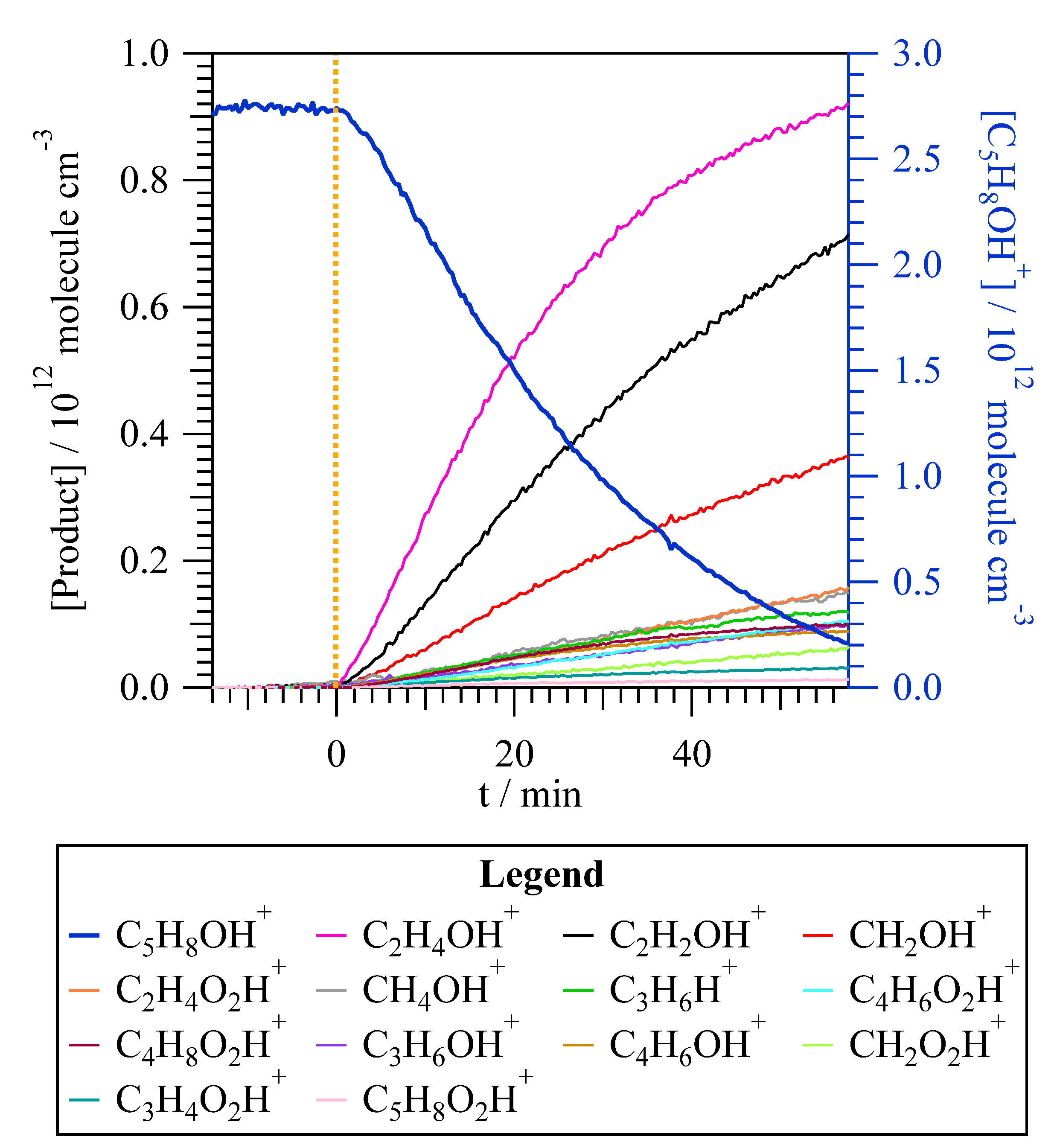
3.2.2. Detection by PTR-ToF-MS
3.2.3. Detection by FTIR Spectroscopy
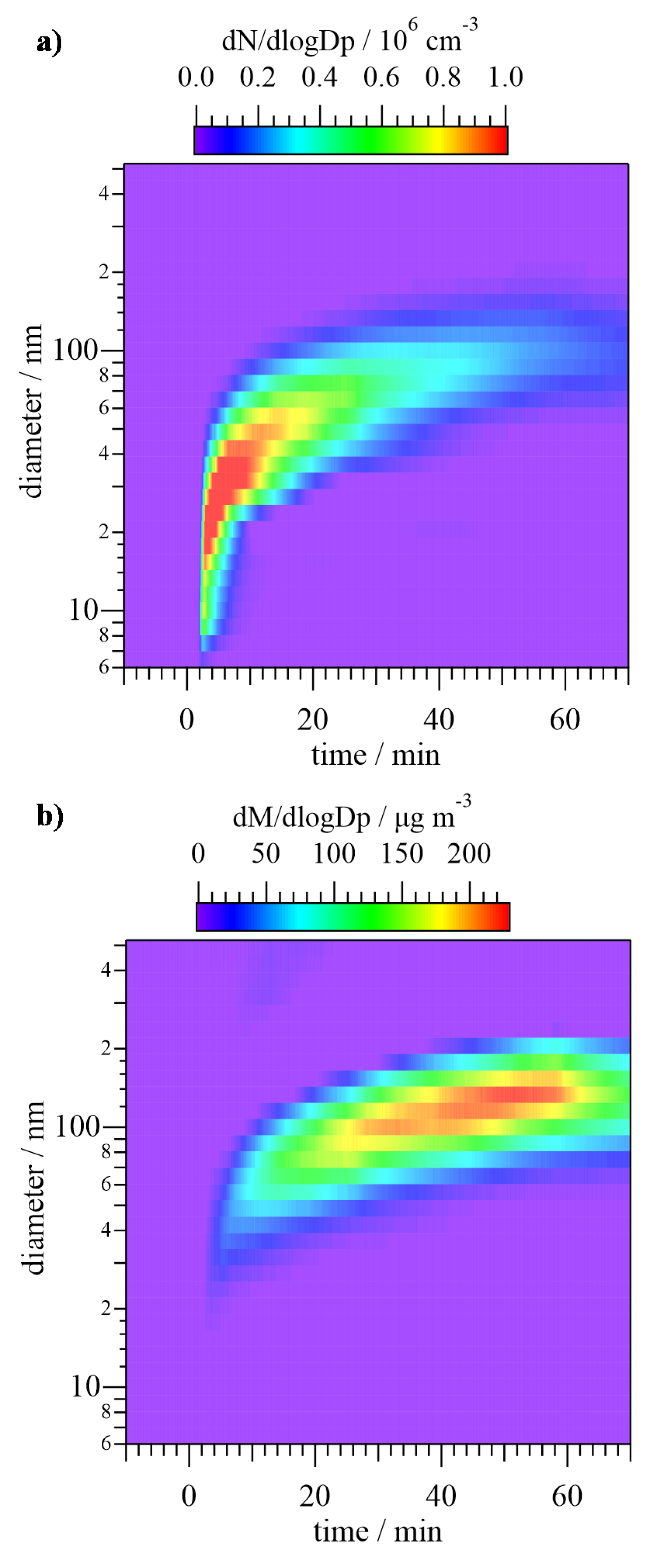
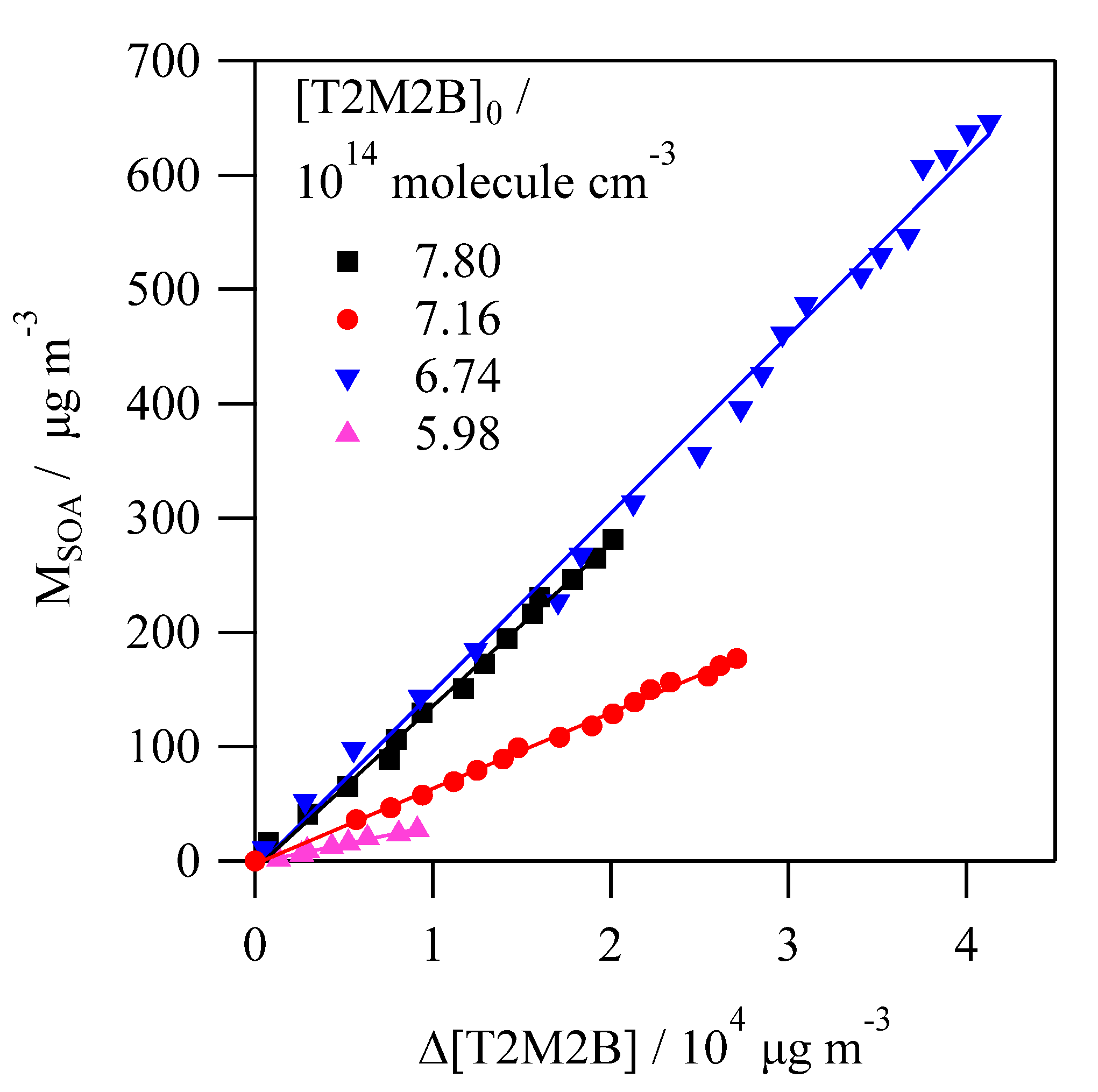
3.3. SOA Formation Study
4. Discussion
4.1. Comparison of the Cl Reactivity with Unsaturated Aldehydes
4.2. Reaction Mechanism to Form Gaseous Products
4.3. SOA Formation Study
4.4. Atmospheric Implications
5. Conclusions
- Acetaldehyde, methylglyoxal, acetic acid, and 2,3-butanedione, detected by the three techniques;
- 3-chloro-2-butanone, detected by SPME/GC-MS and FTIR;
- Formaldehyde, methanol, and ketene, detected by PTR-ToF-MS and FTIR;
- HCl, CO, and ClC(O)H, detected only by FTIR.
Author Contributions
Funding
Conflicts of Interest
References
- Kawamura, K.; Steinberg, S.; Kaplan, I.R. Homologous series of C1–C10 monocarboxylic acids and C1–C6 carbonyls in Los Angeles air and motor vehicle exhausts. Atmos. Environ. 2000, 34, 4175–4191. [Google Scholar] [CrossRef]
- Grosjean, D.; Grosjean, E.; Gertler, A.W. On-Road Emissions of Carbonyls from Light-Duty and Heavy-Duty Vehicles. Environ. Sci. Technol. 2001, 35, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wildt, J.; Kobel, K.; Schuh-Thomas, G.; Heiden, A.C. Emissions of Oxygenated Volatile Organic Compounds from Plants Part II: Emissions of Saturated Aldehydes. J. Atmos. Chem. 2003, 45, 173–196. [Google Scholar] [CrossRef]
- Atkinson, R.; Arey, J. Atmospheric Degradation of Volatile Organic Compounds. Chem. Rev. 2003, 103, 4605–4638. [Google Scholar] [CrossRef]
- Yu, J.; Jeffries, H.E.; Le Lacheur, R.M. Identifying Airborne Carbonyl Compounds in Isoprene Atmospheric Photooxidation Products by Their PFBHA Oximes Using Gas Chromatography/Ion Trap Mass Spectrometry. Environ. Sci. Technol. 1995, 29, 1923–1932. [Google Scholar] [CrossRef]
- Park, J.; Jongsma, C.G.; Zhang, R.; North, S.W. Cyclization reactions in isoprene derived β-hydroxy radicals: Implications for the atmospheric oxidation mechanism. Phys. Chem. Chem. Phys. 2003, 5, 3638–3642. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef]
- Jiménez, E.; Barnes, I. Daytime Atmospheric Chemistry of C4–C7 Saturated and Unsaturated Carbonyl Compounds. In Environment, Energy and Climate Change I: Environmental Chemistry of Pollutants and Wastes; Jiménez, E., Cabañas, B., Lefebvre, G., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 53–103. [Google Scholar]
- Rodríguez, D.; Rodríguez, A.; Notario, A.; Aranda, A.; Díaz-de-Mera, Y.; Martínez, E. Kinetic study of the gas-phase reaction of atomic chlorine with a series of aldehydes. Atmos. Chem. Phys. 2005, 5, 3433–3440. [Google Scholar] [CrossRef] [Green Version]
- Thévenet, R.; Mellouki, A.; Le Bras, G. Kinetics of OH and Cl reactions with a series of aldehydes. Int. J. Chem. Kinet. 2000, 32, 676–685. [Google Scholar] [CrossRef]
- Wang, W.; Ezell, M.J.; Ezell, A.A.; Soskin, G.; Finlayson-Pitts, B.J. Rate constants for the reactions of chlorine atoms with a series of unsaturated aldehydes and ketones at 298 K: Structure and reactivity. Phys. Chem. Chem. Phys. 2002, 4, 1824–1831. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.B.; Barnes, I.; Teruel, M.A. FTIR gas-phase kinetic study of the reactions of Cl atoms with (CH3)2CCHC(O)H and CH3CHCHC(O)OCH3. Chem. Phys. Lett. 2010, 488, 135–139. [Google Scholar] [CrossRef]
- Jiménez, E.; Lanza, B.; Antiñolo, M.; Albaladejo, J. Influence of temperature on the chemical removal of 3-methylbutanal, trans-2-methyl-2-butenal, and 3-methyl-2-butenal by OH radicals in the troposphere. Atmos. Environ. 2009, 43, 4043–4049. [Google Scholar] [CrossRef]
- Sato, K.; Klotz, B.; Taketsugu, T.; Takayanagi, T. Kinetic measurements for the reactions of ozone with crotonaldehyde and its methyl derivatives and calculations of transition-state theory. Phys. Chem. Chem. Phys. 2004, 6, 3969–3976. [Google Scholar] [CrossRef]
- Grosjean, D.; Williams, E.L. Environmental persistence of organic compounds estimated from structure-reactivity and linear free-energy relationships. Unsaturated aliphatics. Atmos. Environ. Part A Gen. Top. 1992, 26, 1395–1405. [Google Scholar] [CrossRef]
- Jang, M.; Czoschke, N.M.; Lee, S.; Kamens, R.M. Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions. Science 2002, 298, 814. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Chan, M.N.; Surratt, J.D.; Chhabra, P.S.; Loza, C.L.; Crounse, J.D.; Yee, L.D.; Flagan, R.C.; Wennberg, P.O.; Seinfeld, J.H. Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation. Atmos. Chem. Phys. 2010, 10, 7169–7188. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, B.; Jimenez, E.; Moreno, A.; Soto, A.; Antinolo, M.; Albaladejo, J. Atmospheric fate of hydrofluoroolefins, CxF2x+1CH=CH2 (x = 1,2,3,4 and 6): Kinetics with Cl atoms and products. Chemosphere 2017, 167, 330–343. [Google Scholar] [CrossRef]
- Antiñolo, M.; Olmo, R.D.; Bravo, I.; Albaladejo, J.; Jiménez, E. Tropospheric fate of allyl cyanide (CH2CHCH2CN): Kinetics, reaction products and secondary organic aerosol formation. Atmos. Environ. 2019, 219, 117041. [Google Scholar] [CrossRef]
- Aschmann, S.M.; Atkinson, R. Rate constants for the gas-phase reactions of alkanes with Cl atoms at 296 ± 2 K. Int. J. Chem. Kinet. 1995, 27, 613–622. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S.; Apel, E.C.; Riemer, D.D.; Paulson, S.E. Rate coefficients and mechanisms of the reaction of cl-atoms with a series of unsaturated hydrocarbons under atmospheric conditions. Int. J. Chem. Kinet. 2003, 35, 334–353. [Google Scholar] [CrossRef]
- Wallington, T.J.; Ball, J.C.; Straccia, A.M.; Hurley, M.D.; Kaiser, E.W.; Dill, M.; Schneider, W.F.; Bilde, M. Kinetics and mechanism of the reaction of Cl atoms with CH2 CO (Ketene). Int. J. Chem. Kinet. 1996, 28, 627–635. [Google Scholar] [CrossRef]
- Nielsen, O.J.; University of Copenhangen, København, Denmark. Personal communication, 2019.
- Eurochamp Data Center. 2017. Available online: https://data.eurochamp.org/ (accessed on 1 June 2020).
- Pankow, J.F. An absorption model of gas/particle partitioning of organic compounds in the atmosphere. Atmos. Environ. 1994, 28, 185–188. [Google Scholar] [CrossRef]
- Pankow, J.F. An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol. Atmos. Environ. 1994, 28, 189–193. [Google Scholar] [CrossRef]
- Odum, J.R.; Hoffmann, T.; Bowman, F.; Collins, D.; Flagan, R.C.; Seinfeld, J.H. Gas/particle partitioning and secondary organic aerosol yields. Environ. Sci. Technol. 1996, 30, 2580–2585. [Google Scholar] [CrossRef]
- Hasson, A.S.; Tyndall, G.S.; Orlando, J.J.; Singh, S.; Hernandez, S.Q.; Campbell, S.; Ibarra, Y. Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals. J. Phys. Chem. A 2012, 116, 6264–6281. [Google Scholar] [CrossRef]
- Magneron, I.; Thévenet, R.; Mellouki, A.; Le Bras, G.; Moortgat, G.K.; Wirtz, K. A Study of the Photolysis and OH-initiated Oxidation of Acrolein and trans-Crotonaldehyde. J. Phys. Chem. A 2002, 106, 2526–2537. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S. Mechanisms for the Reactions of OH with Two Unsaturated Aldehydes: Crotonaldehyde and Acrolein. J. Phys. Chem. A 2002, 106, 12252–12259. [Google Scholar] [CrossRef]
- Tuazon, E.C.; Aschmann, S.M.; Nishino, N.; Arey, J.; Atkinson, R. Kinetics and products of the OH radical-initiated reaction of 3-methyl-2-butenal. Phys. Chem. Chem. Phys. 2005, 7, 2298–2304. [Google Scholar] [CrossRef]
- Maricq, M.M.; Ball, J.C.; Straccia, A.M.; Szente, J.J. A diode laser study of the Cl + CH3CO reaction. Int. J. Chem. Kinet. 1997, 29, 421–429. [Google Scholar] [CrossRef]
- Cai, X.; Griffin, R.J. Secondary aerosol formation from the oxidation of biogenic hydrocarbons by chlorine atoms. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Jiang, J.; Lu, Z.; Wang, K.; Li, J.; Hao, J. Effects of Inorganic Seeds on Secondary Organic Aerosol (SOA) Formation. In Atmospheric Aerosols—Regional Characteristics—Chemistry and Physics; Abdul-Razzak, H., Ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Singh, H.B.; Thakur, A.N.; Chen, Y.E.; Kanakidou, M. Tetrachloroethylene as an indicator of low Cl atom concentrations in the troposphere. Geophys. Res. Lett. 1996, 23, 1529–1532. [Google Scholar] [CrossRef]
- Spicer, C.W.; Chapman, E.G.; Finlayson-Pitts, B.J.; Plastridge, R.A.; Hubbe, J.M.; Fast, J.D.; Berkowitz, C.M. Unexpectedly high concentrations of molecular chlorine in coastal air. Nature 1998, 394, 353–356. [Google Scholar] [CrossRef]
- Lanza, B.; Jiménez, E.; Ballesteros, B.; Albaladejo, J. Absorption cross section determination of biogenic C5-aldehydes in the actinic region. Chem. Phys. Lett. 2008, 454, 184–189. [Google Scholar] [CrossRef]
- Sanhueza, E. Hydrochloric acid from chlorocarbons: A significant global source of background rain acidity. Tellus B Chem. Phys. Meteorol. 2001, 53, 122–132. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kw/10−6 s−1 | khν/10−4 s−1 | kCl2/10−20 cm3 molecule−1 s−1 |
|---|---|---|---|
| Trans-2-methyl-2-butenal | 8.40 ± 5.33 | 8.33 ± 5.60 | - |
| Cyclohexane | 0.400 ± 0.417 | - | - |
| Isoprene | 3.78 ± 1.65 | - | 2.70 ± 0.15 |
| Reference | k/kRef | kRef/10−10 cm3 molecule−1 s−1 | k/10−10 cm3 molecule−1 s−1 |
|---|---|---|---|
| Cyclohexane | 0.894 ± 0.030 | 3.08 ± 0.12 1 | 2.75 ± 0.14 |
| Isoprene | 0.491 ± 0.014 | 4.35 ± 0.58 2 | 2.14 ± 0.29 |
| Average | 2.45 ± 0.32 |
| [T2M2B]0/1014 molecule cm−3 | [Cl2]0/1014 molecule cm−3 | MSOA/μg m−3 | YSOA (%) |
|---|---|---|---|
| 5.62 | 4.46 | 27.4 | 0.26 ± 0.01 |
| 6.79 | 2.54 | 52.9 | 0.27 ± 0.01 |
| 5.99 | 2.67 | 60.9 | 0.33 ± 0.01 |
| 7.17 | 5.96 | 191 | 0.68 ± 0.01 |
| 6.31 | 6.93 | 244 | 0.96 ± 0.01 |
| 7.79 | 9.53 | 441 | 1.42 ± 0.03 |
| 7.58 | 12.6 | 491 | 1.49 ± 0.04 |
| 7.39 | 15.0 | 580 | 1.44 ± 0.02 |
| 6.74 | 17.0 | 764 | 1.56 ± 0.03 |
| 7.10 | 19.9 | 863 | 1.65 ± 0.03 |
| Unsaturated Aldehyde | k/10−10 cm3 molecule−1 s−1 | Technique | Reference | |
|---|---|---|---|---|
| Linear | E-CH3CH=CHC(O)H | 2.60 ± 0.04 | GC-FID | [10] |
| E-CH3CH2CH=CHC(O)H | 1.31 ± 0.19 | GC-MS | [9] | |
| E-CH3(CH2)2CH=CHC(O)H | 1.92 ± 0.22 | GC-MS | [9] | |
| E-CH3(CH2)3CH=CHC(O)H | 2.40 ± 0.29 | GC-MS | [9] | |
| Branched | E-CH3CH=C(CH3)C(O)H | 2.45 ± 0.32 | FTIR | This research |
| (CH3)2C=CHC(O)H | 2.48 ± 0.71 | FTIR | [12] | |
| CH2=C(CH3)C(O)H | 2.9 ± 0.8 | FTIR and GC-FID | [11] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antiñolo, M.; Asensio, M.; Albaladejo, J.; Jiménez, E. Gas-Phase Reaction of trans-2-Methyl-2-butenal with Cl: Kinetics, Gaseous Products, and SOA Formation. Atmosphere 2020, 11, 715. https://doi.org/10.3390/atmos11070715
Antiñolo M, Asensio M, Albaladejo J, Jiménez E. Gas-Phase Reaction of trans-2-Methyl-2-butenal with Cl: Kinetics, Gaseous Products, and SOA Formation. Atmosphere. 2020; 11(7):715. https://doi.org/10.3390/atmos11070715
Chicago/Turabian StyleAntiñolo, María, María Asensio, José Albaladejo, and Elena Jiménez. 2020. "Gas-Phase Reaction of trans-2-Methyl-2-butenal with Cl: Kinetics, Gaseous Products, and SOA Formation" Atmosphere 11, no. 7: 715. https://doi.org/10.3390/atmos11070715
APA StyleAntiñolo, M., Asensio, M., Albaladejo, J., & Jiménez, E. (2020). Gas-Phase Reaction of trans-2-Methyl-2-butenal with Cl: Kinetics, Gaseous Products, and SOA Formation. Atmosphere, 11(7), 715. https://doi.org/10.3390/atmos11070715