Airborne Bacterial and Eukaryotic Community Structure across the United Kingdom Revealed by High-Throughput Sequencing


 ,
, 
 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experiments
2.1. Sample Collection
2.2. DNA Extraction, PCR, and Sequencing
2.3. Sequence Analysis
2.4. Statistical Analysis
3. Results and Discussion
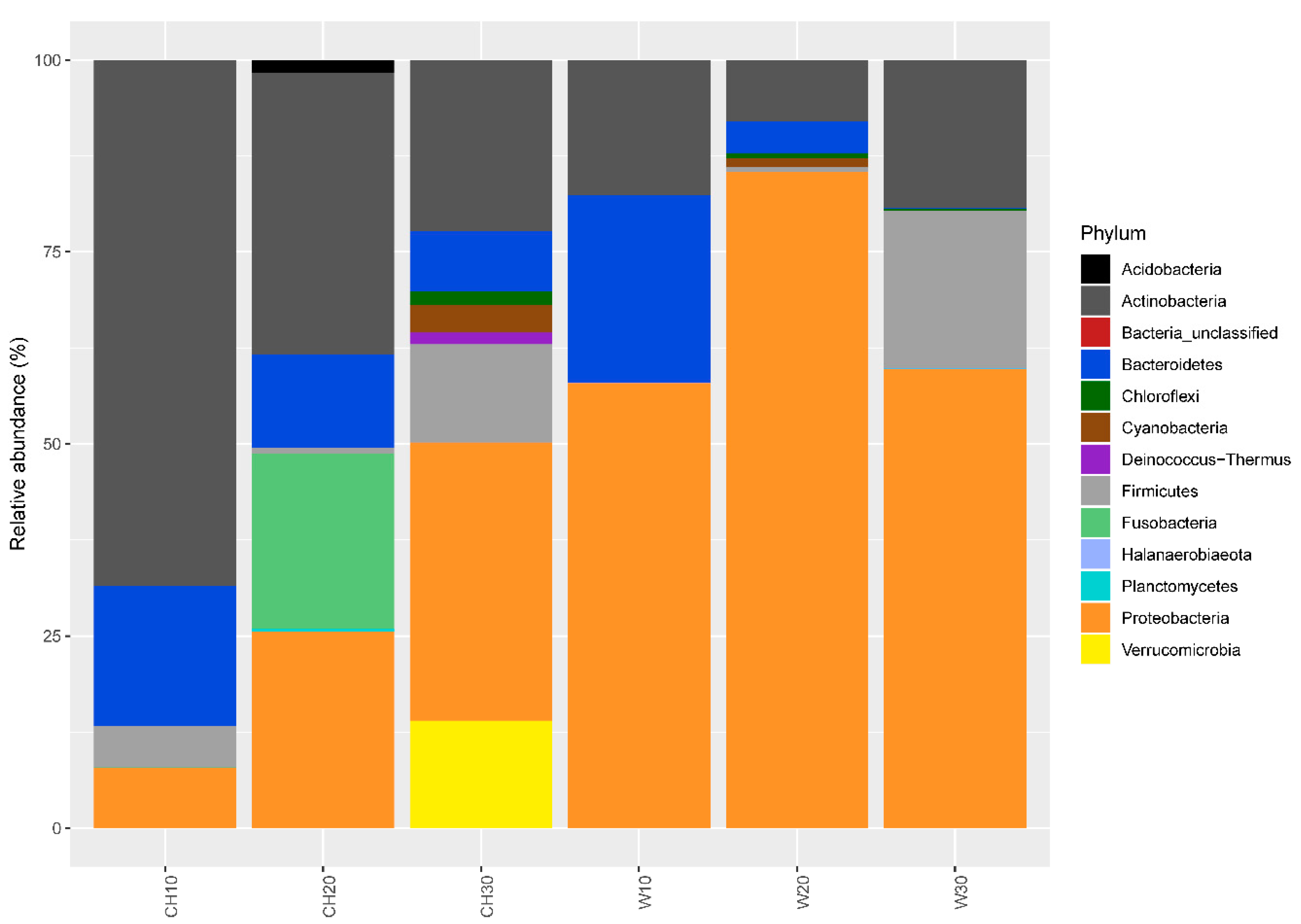
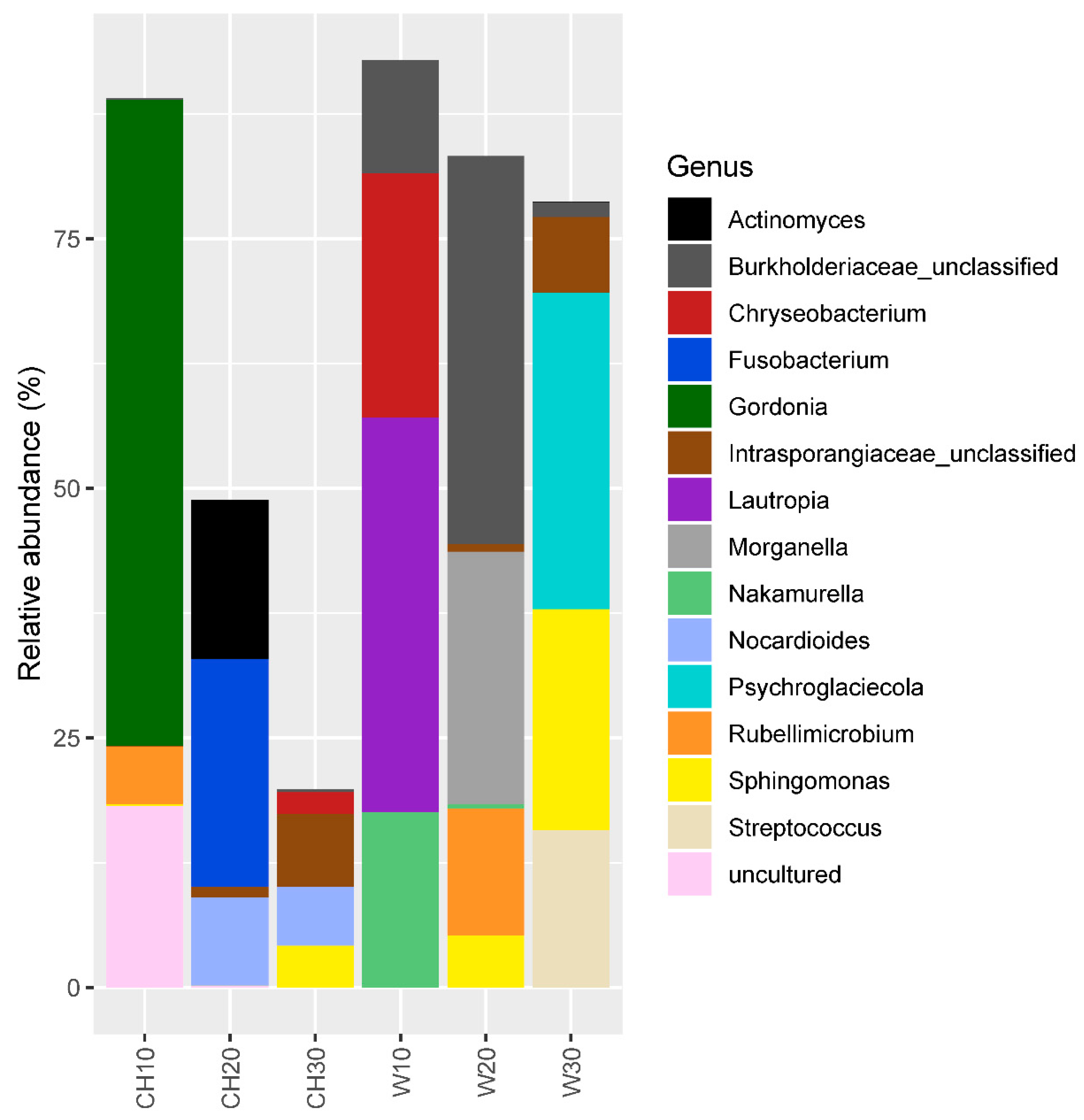
3.1. Bacterial Community Structure and Diversity
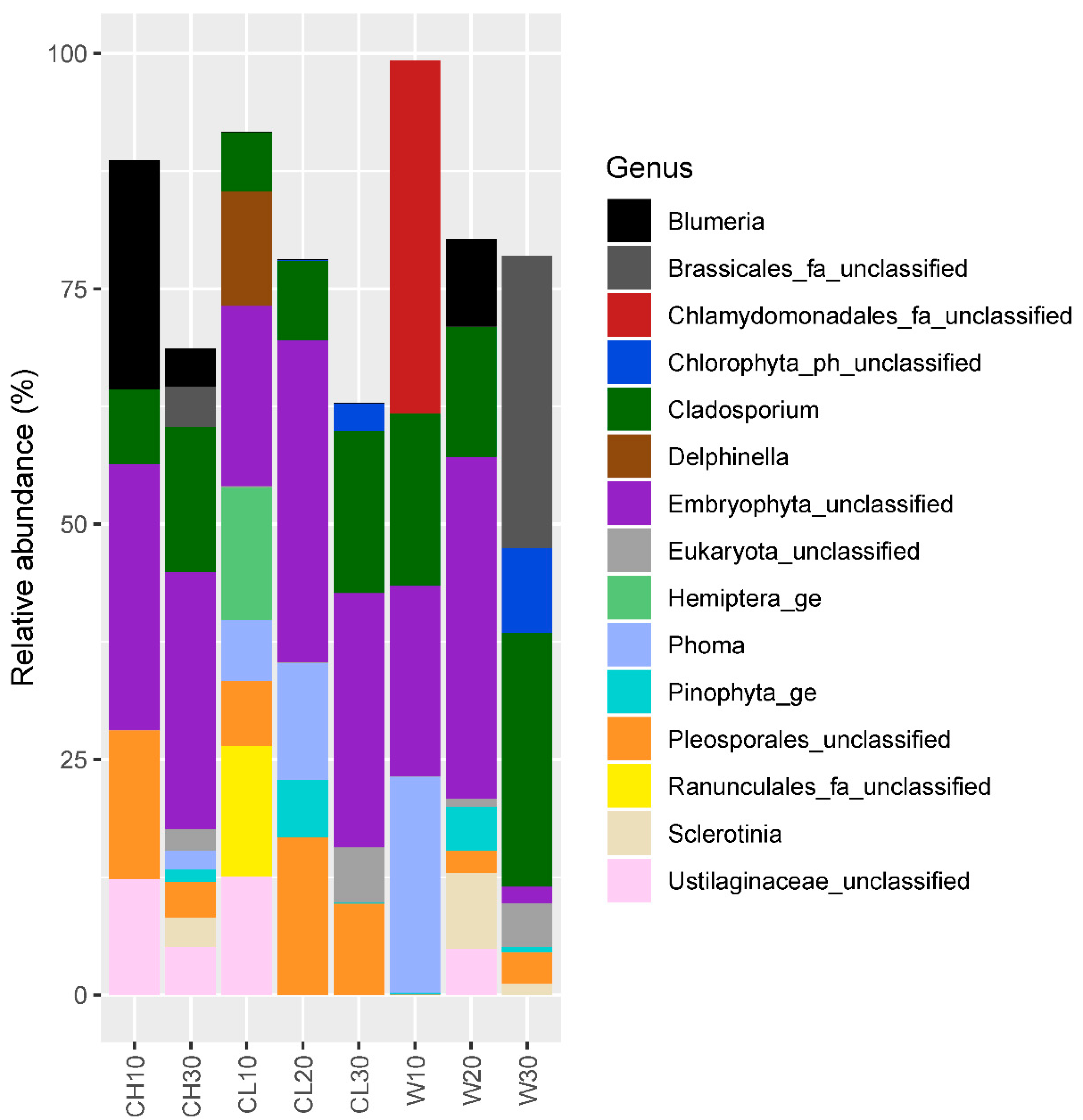
3.2. Eukaryotic Community Structure and Diversity
3.3. Possible Limitation of the Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yooseph, S.; Andrews-Pfannkoch, C.; Tenney, A.; McQuaid, J.; Williamson, S.; Thiagarajan, M.; Brami, D.; Zeigler-Allen, L.; Hoffman, J.; Goll, J.B.; et al. A Metagenomic Framework for the Study of Airborne Microbial Communities. PLoS ONE 2013, 8, e81862. [Google Scholar] [CrossRef]
- Gupta, R.; Sheikh, A.; Strachan, D.P.; Anderson, H.R. Burden of allergic disease in the UK: Secondary analyses of national databases. Clin. Exp. Allergy 2004, 34, 520–526. [Google Scholar] [CrossRef]
- Ukhanova, O.; Bogomolova, E. Chapter 2—Airborne Allergens. In Allergic Diseases: New Insights; Celso, P., Ed.; IntechOpen: Rijeka, Croatia, 2015; pp. 35–68. [Google Scholar]
- McInnes, R.N.; Hemming, D.; Burgess, P.; Lyndsay, D.; Osborne, N.J.; Skjøth, C.A.; Thomas, S.; Vardoulakis, S. Mapping allergenic pollen vegetation in UK to study environmental exposure and human health. Sci. Total Environ. 2017, 599, 483–499. [Google Scholar] [CrossRef]
- Lockey, R.F.; Ledford, D.K. Allergens and Allergen Immunotherapy: Subcutaneous, Sublingual and Oral, 6th ed.; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Levetin, E.; Horner, W.E.; Scott, J.A.; Barnes, C.; Baxi, S.; Chew, G.L.; Grimes, C.; Horner, W.E.; Kennedy, K.; Larenas-Linnemann, D.; et al. Taxonomy of Allergenic Fungi. J. Allergy Clin. Immunol. Pract. 2016, 4, 375–385.e371. [Google Scholar] [CrossRef]
- Simon-Nobbe, B.; Denk, U.; Pöll, V.; Rid, R.; Breitenbach, M. The Spectrum of Fungal Allergy. Int. Arch. Allergy Immunol. 2008, 145, 58–86. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, C. Airborne Infectious Microorganisms. Encycl. Microbiol. 2019, 52. [Google Scholar] [CrossRef]
- Chowell, G.; Ammon, C.E.; Hengartner, N.W.; Hyman, J.M. Transmission dynamics of the great influenza pandemic of 1918 in Geneva, Switzerland: Assessing the effects of hypothetical interventions. J. Theor. Biol. 2006, 241, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Brosseau, L.M. Aerosol Transmission of Infectious Disease. J. Occup. Environ. Med. 2015, 57, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Webster, R.G. The Influenza Virus Enigma. Cell 2009, 136, 402–410. [Google Scholar] [CrossRef]
- Tellier, R.; Li, Y.; Cowling, B.J.; Tang, J.W. Recognition of aerosol transmission of infectious agents: A commentary. BMC Infect. Dis. 2019, 19, 101. [Google Scholar] [CrossRef]
- Smets, W.; Moretti, S.; Denys, S.; Lebeer, S. Airborne bacteria in the atmosphere: Presence, purpose, and potential. Atmos. Environ. 2016, 139, 214–221. [Google Scholar] [CrossRef]
- Hirst, J.M.; Stedman, O.J. The epidemiology of phytophthora infestans. Ann. Appl. Biol. 1960, 48, 489–517. [Google Scholar] [CrossRef]
- Skelsey, P.; Kessel, G.J.T.; Holtslag, A.A.M.; Moene, A.F.; van der Werf, W. Regional spore dispersal as a factor in disease risk warnings for potato late blight: A proof of concept. Agric. For. Meteorol. 2009, 149, 419–430. [Google Scholar] [CrossRef]
- Marianelli, C.; Amato, B.; Boniotti, M.B.; Vitale, M.; Pruiti Ciarello, F.; Pacciarini, M.L.; Di Marco Lo Presti, V. Genotype diversity and distribution of Mycobacterium bovis from livestock in a small, high-risk area in northeastern Sicily, Italy. PLoS Negl. Trop. Dis. 2019, 13, e0007546. [Google Scholar] [CrossRef] [PubMed]
- Torres-Gonzalez, P.; Cervera-Hernandez, M.E.; Martinez-Gamboa, A.; Garcia-Garcia, L.; Cruz-Hervert, L.P.; Bobadilla-del Valle, M.; Ponce-de Leon, A.; Sifuentes-Osornio, J. Human tuberculosis caused by Mycobacterium bovis: A retrospective comparison with Mycobacterium tuberculosis in a Mexican tertiary care centre, 2000–2015. BMC Infect. Dis. 2016, 16, 657. [Google Scholar] [CrossRef] [PubMed]
- Aranaz, A.; de Juan, L.; Montero, N.; Sánchez, C.; Galka, M.; Delso, C.; Álvarez, J.; Romero, B.; Bezos, J.; Vela, A.I.; et al. Bovine Tuberculosis (Mycobacterium bovis) in Wildlife in Spain. J. Clin. Microbiol. 2004, 42, 2602–2608. [Google Scholar] [CrossRef]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef]
- Hirano, S.S.; Upper, C.D. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 2000, 64, 624–653. [Google Scholar] [CrossRef]
- Ruske, S.; Topping, D.O.; Foot, V.E.; Kaye, P.H.; Stanley, W.R.; Crawford, I.; Morse, A.P.; Gallagher, M.W. Evaluation of machine learning algorithms for classification of primary biological aerosol using a new UV-LIF spectrometer. Atmos. Meas. Tech. 2017, 10, 695–708. [Google Scholar] [CrossRef]
- Huffman, J.A.; Perring, A.E.; Savage, N.J.; Clot, B.; Crouzy, B.; Tummon, F.; Shoshanim, O.; Damit, B.; Schneider, J.; Sivaprakasam, V.; et al. Real-time sensing of bioaerosols: Review and current perspectives. Aerosol Sci. Technol. 2020, 54, 465–495. [Google Scholar] [CrossRef]
- Li, H.; Zhou, X.-Y.; Yang, X.-R.; Zhu, Y.-G.; Hong, Y.-W.; Su, J.-Q. Spatial and seasonal variation of the airborne microbiome in a rapidly developing city of China. Sci. Total Environ. 2019, 665, 61–68. [Google Scholar] [CrossRef]
- Núñez, A.; Moreno, D.A. The Differential Vertical Distribution of the Airborne Biological Particles Reveals an Atmospheric Reservoir of Microbial Pathogens and Aeroallergens. Microb. Ecol. 2020. [Google Scholar] [CrossRef]
- DeLeon-Rodriguez, N.; Lathem, T.L.; Rodriguez-R, L.M.; Barazesh, J.M.; Anderson, B.E.; Beyersdorf, A.J.; Ziemba, L.D.; Bergin, M.; Nenes, A.; Konstantinidis, K.T. Microbiome of the upper troposphere: Species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc. Natl. Acad. Sci. USA 2013, 110, 2575–2580. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Ravichandar, J.D.; Jain, S.; Griffin, D.W.; Yu, H.; Tan, Q.; Thissen, J.; Lusby, T.; Nicoll, P.; Shedler, S.; et al. Airborne Bacteria in Earth’s Lower Stratosphere Resemble Taxa Detected in the Troposphere: Results from a New NASA Aircraft Bioaerosol Collector (ABC). Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Hara, K.; Iwata, A.; Lee, K.C.; Kawai, K.; Kai, K.; Kobayashi, F.; Pointing, S.B.; Archer, S.; Hasegawa, H.; et al. Variations in airborne bacterial communities at high altitudes over the Noto Peninsula (Japan) in response to Asian dust events. Atmos. Chem. Phys. 2017, 17, 11877–11897. [Google Scholar] [CrossRef]
- Forde, E.; Gallagher, M.; Foot, V.; Sarda-Esteve, R.; Crawford, I.; Kaye, P.; Stanley, W.; Topping, D. Characterisation and source identification of biofluorescent aerosol emissions over winter and summer periods in the United Kingdom. Atmos. Chem. Phys. 2019, 19, 1665–1684. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.-W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19, 21–31. [Google Scholar] [CrossRef]
- Lane, D. Nucleic Acid Technologies in Bacterial Systematic; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Medlin, L.; Elwood, H.J.; Stickel, S.; Sogin, M.L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 1988, 71, 491–499. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- MiSeq SOP. Available online: https://mothur.org/wiki/miseq_sop/ (accessed on 1 September 2019).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Westcott, S.L.; Schloss, P.D. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 46, D8–D13. [Google Scholar] [CrossRef]
- Clarke, K.R.; Clarke, K.; Gorley, K.; Clarke, K.; Gorley, R. PRIMER V6: User Manual/Tutorial; PRIMER-E Ltd.: Plymouth, UK, 2006. [Google Scholar]
- Šmilauer, P.; Lepš, J. Multivariate Analysis of Ecological Data Using CANOCO 5; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2016, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Sowani, H.; Kulkarni, M.; Zinjarde, S. An insight into the ecology, diversity and adaptations of Gordonia species. Crit. Rev. Microbiol. 2018, 44, 393–413. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Kang, Y.; Yazawa, K.; Gonoi, T.; Kamei, K.; Mikami, Y. Characterization of Clinical Isolates of Gordonia Species in Japanese Clinical Samples during 1998–2008. Mycopathologia 2009, 168, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Richet, H.M.; Craven, P.C.; Brown, J.M.; Lasker, B.A.; Cox, C.D.; McNeil, M.M.; Tice, A.D.; Jarvis, W.R.; Tablan, O.C. A Cluster of Rhodococcus (Gordona) Bronchialis Sternal-Wound Infections after Coronary-Artery Bypass Surgery. N. Engl. J. Med. 1991, 324, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Wang, C.Y.; Liu, C.Y.; Tan, C.K.; Lin, S.H.; Liao, C.H.; Chou, C.H.; Huang, Y.T.; Lin, H.I.; Hsueh, P.R. Infections caused by Gordonia species at a medical centre in Taiwan, 1997 to 2008. Clin. Microbiol. Infect. 2010, 16, 1448–1453. [Google Scholar] [CrossRef]
- Verma, P.; Brown, J.M.; Nunez, V.H.; Morey, R.E.; Steigerwalt, A.G.; Pellegrini, G.J.; Kessler, H.A. Native valve endocarditis due to Gordonia polyisoprenivorans: Case report and review of literature of bloodstream infections caused by Gordonia species. J. Clin. Microbiol. 2006, 44, 1905–1908. [Google Scholar] [CrossRef]
- Kempf, V.A.J.; Schmalzing, M.; Yassin, A.F.; Schaal, K.P.; Baumeister, D.; Arenskötter, M.; Steinbüchel, A.; Autenrieth, I.B. Gordonia polyisoprenivorans Septicemia in a Bone Marrow Transplant Patient. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 226–228. [Google Scholar] [CrossRef]
- Kang, Y.-Q.; Ming, H.; Gonoi, T.; Chen, Y.; Cao, Y.; Wang, Y.-Y.; Cheng, J.; Koga, T.; Mikami, Y.; Li, W.-J. Gordonia iterans sp. nov., isolated from a patient with pneumonia. Int. J. Syst. Evol. Microbiol. 2014, 64, 3520–3525. [Google Scholar] [CrossRef]
- Signat, B.; Roques, C.; Poulet, P.; Duffaut, D. Fusobacterium nucleatum in periodontal health and disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar]
- Nagaraja, T.G.; Narayanan, S.K.; Stewart, G.C.; Chengappa, M.M. Fusobacterium necrophorum infections in animals: Pathogenesis and pathogenic mechanisms. Anaerobe 2005, 11, 239–246. [Google Scholar] [CrossRef]
- Bradshaw, D.J.; Marsh, P.D.; Watson, G.K.; Allison, C. Role of Fusobacterium nucleatum and Coaggregation in Anaerobe Survival in Planktonic and Biofilm Oral Microbial Communities during Aeration. Infect. Immun. 1998, 66, 4729–4732. [Google Scholar] [CrossRef]
- Löndahl, J. Physical and Biological Properties of Bioaerosols. In Bioaerosol Detection Technologies; Jonsson, P., Olofsson, G., Tjärnhage, T., Eds.; Springer: New York, NY, USA, 2014; pp. 33–48. [Google Scholar] [CrossRef]
- Barson, W.J.; Marcon, M.J. 144—Proteus, Providencia, and Morganella Species. In Principles and Practice of Pediatric Infectious Diseases, 4th ed.; Long, S.S., Ed.; Content Repository: London, UK, 2012; pp. 811–812.e812. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, J.; Hu, Q.; Rao, X. Morganella morganii, a non-negligent opportunistic pathogen. Int. J. Infect. Dis. 2016, 50, 10–17. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, C.M.; Brenner, F.W.; Miller, J.M. Classification, Identification, and Clinical Significance of Proteus, Providencia, and Morganella. Clin. Microbiol. Rev. 2000, 13, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Failor, K.C.; Schmale, D.G., 3rd; Vinatzer, B.A.; Monteil, C.L. Ice nucleation active bacteria in precipitation are genetically diverse and nucleate ice by employing different mechanisms. ISME J. 2017, 11, 2740–2753. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, V.; Gandolfi, I.; Ambrosini, R.; Bestetti, G.; Innocente, E.; Rampazzo, G.; Franzetti, A. Temporal variability and effect of environmental variables on airborne bacterial communities in an urban area of Northern Italy. Appl. Microbiol. Biotechnol. 2013, 97, 6561–6570. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Q.; Deng, Y.; Wang, Y.; Wang, X.; Zhang, H.; Sun, X.; Ouyang, Z. Meteorological factors had more impact on airborne bacterial communities than air pollutants. Sci. Total Environ. 2017, 601, 703–712. [Google Scholar] [CrossRef]
- Ehrlich, R.; Miller, S.; Walker, R.L. Relationship between Atmospheric Temperature and Survival of Airborne Bacteria. Appl. Microbiol. 1970, 19, 245–249. [Google Scholar] [CrossRef]
- Després, V.; Huffman, J.A.; Burrows, S.M.; Hoose, C.; Safatov, A.; Buryak, G.; Fröhlich-Nowoisky, J.; Elbert, W.; Andreae, M.; Pöschl, U.; et al. Primary biological aerosol particles in the atmosphere: A review. Tellus B: Chem. Phys. Meteorol. 2012, 64, 15598. [Google Scholar] [CrossRef]
- Stewart, G.A.; Robinson, C. Chapter 4—Indoor and Outdoor Allergens and Pollutants. In Middleton’s Allergy Essentials; O’Hehir, R.E., Holgate, S.T., Sheikh, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 73–116. [Google Scholar] [CrossRef]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus Cladosporium. Stud. Mycol. 2012, 72, 1–401. [Google Scholar] [CrossRef]
- Sandoval-Denis, M.; Sutton, D.A.; Martin-Vicente, A.; Cano-Lira, J.F.; Wiederhold, N.; Guarro, J.; Gené, J. Cladosporium Species Recovered from Clinical Samples in the United States. J. Clin. Microbiol. 2015, 53, 2990–3000. [Google Scholar] [CrossRef]
- Mercier, E.; Peters, I.R.; Billen, F.; Battaille, G.; Clercx, C.; Day, M.J.; Peeters, D. Potential role of Alternaria and Cladosporium species in canine lymphoplasmacytic rhinitis. J. Small Anim. Pract. 2013, 54, 179–183. [Google Scholar] [CrossRef]
- Aveskamp, M.M.; De Gruyter, J.; Crous, P.W. Biology and recent developments in the systematics of Phoma, a complex genus of major quarantine significance. Fungal Divers. 2008, 31, 1–18. [Google Scholar]
- Bennett, A.; Ponder, M.M.; Garcia-Diaz, J. Phoma Infections: Classification, Potential Food Sources, and Its Clinical Impact. Microorganisms 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Pomés, A.; Davies, J.M.; Gadermaier, G.; Hilger, C.; Holzhauser, T.; Lidholm, J.; Lopata, A.L.; Mueller, G.A.; Nandy, A.; Radauer, C.; et al. WHO/IUIS Allergen Nomenclature: Providing a common language. Mol. Immunol. 2018, 100, 3–13. [Google Scholar] [CrossRef] [PubMed]
- McGovern, R.J.; Horita, H.; Stiles, C.M.; Seijo, T.E. Host Range of Itersonilia perplexans and Management of Itersonilia Petal Blight of China Aster. Plant. Health Prog. 2006, 7, 7. [Google Scholar] [CrossRef]
- Talgø, V.; Skage, J.-O.; Steffenrem, A.; Junker, C.; Eikemo, H.; Brurberg, M.B.; Johnskås, O.R. Delphinella Shoot Blight on Abies lasiocarpa Provenances in Norway. Forests 2016, 7, 7. [Google Scholar] [CrossRef]
- Jiang, D.; Fu, Y.; Guoqing, L.; Ghabrial, S.A. Chapter Eight—Viruses of the Plant Pathogenic Fungus Sclerotinia sclerotiorum. In Advances in Virus Research; Ghabrial, S.A., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 86, pp. 215–248. [Google Scholar]
- Feldbrügge, M.; Kellner, R.; Schipper, K. The biotechnological use and potential of plant pathogenic smut fungi. Appl. Microbiol. Biotechnol. 2013, 97, 3253–3265. [Google Scholar] [CrossRef]
- Geiser, E.; Wiebach, V.; Wierckx, N.; Blank, L.M. Prospecting the biodiversity of the fungal family Ustilaginaceae for the production of value-added chemicals. Fungal Biol. Biotechnol. 2014, 1, 1–10. [Google Scholar] [CrossRef]
- Inuma, T.; Khodaparast, S.A.; Takamatsu, S. Multilocus phylogenetic analyses within Blumeria graminis, a powdery mildew fungus of cereals. Mol. Phylogenetics Evol. 2007, 44, 741–751. [Google Scholar] [CrossRef]
- Zhang, Y.; Crous, P.W.; Schoch, C.L.; Hyde, K.D. Pleosporales. Fungal Divers. 2012, 53, 1–221. [Google Scholar] [CrossRef]
- Xu, J.-X.; Zhang, D.-S.; Li, L.-H. Seasonal variations of airborne pollen in Beijing, China and their relationships with meteorological factors. Acta Ecol. Sin. 2012, 32, 202–208. [Google Scholar] [CrossRef]
- Barboni, D.; Bonnefille, R.; Prasad, S.; Ramesh, B.R. Variation in modern pollen from tropical evergreen forests and the monsoon seasonality gradient in SW India. J. Veg. Sci. 2003, 14, 551–562. [Google Scholar] [CrossRef]
- Priyamvada, H.; Singh, R.K.; Akila, M.; Ravikrishna, R.; Verma, R.S.; Gunthe, S.S. Seasonal variation of the dominant allergenic fungal aerosols—One year study from southern Indian region. Sci. Rep. 2017, 7, 11171. [Google Scholar] [CrossRef] [PubMed]
- Ezike, D.N.; Nnamani, C.V.; Ogundipe, O.T.; Adekanmbi, O.H. Airborne pollen and fungal spores in Garki, Abuja (North-Central Nigeria). Aerobiologia 2016, 32, 697–707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Crawford, I.; Lloyd, J.R.; Robinson, C.H.; Boothman, C.; Bower, K.; Gallagher, M.; Allen, G.; Topping, D. Airborne Bacterial and Eukaryotic Community Structure across the United Kingdom Revealed by High-Throughput Sequencing. Atmosphere 2020, 11, 802. https://doi.org/10.3390/atmos11080802
Song H, Crawford I, Lloyd JR, Robinson CH, Boothman C, Bower K, Gallagher M, Allen G, Topping D. Airborne Bacterial and Eukaryotic Community Structure across the United Kingdom Revealed by High-Throughput Sequencing. Atmosphere. 2020; 11(8):802. https://doi.org/10.3390/atmos11080802
Chicago/Turabian StyleSong, Hokyung, Ian Crawford, Jonathan R. Lloyd, Clare H. Robinson, Christopher Boothman, Keith Bower, Martin Gallagher, Grant Allen, and David Topping. 2020. "Airborne Bacterial and Eukaryotic Community Structure across the United Kingdom Revealed by High-Throughput Sequencing" Atmosphere 11, no. 8: 802. https://doi.org/10.3390/atmos11080802
APA StyleSong, H., Crawford, I., Lloyd, J. R., Robinson, C. H., Boothman, C., Bower, K., Gallagher, M., Allen, G., & Topping, D. (2020). Airborne Bacterial and Eukaryotic Community Structure across the United Kingdom Revealed by High-Throughput Sequencing. Atmosphere, 11(8), 802. https://doi.org/10.3390/atmos11080802