
Effect of Water Molecule on the Complete Series Reactions of Chlorothiobenzenes with H/·OH: A Theoretical Study

Abstract
:1. Introduction
2. Results and Discussion
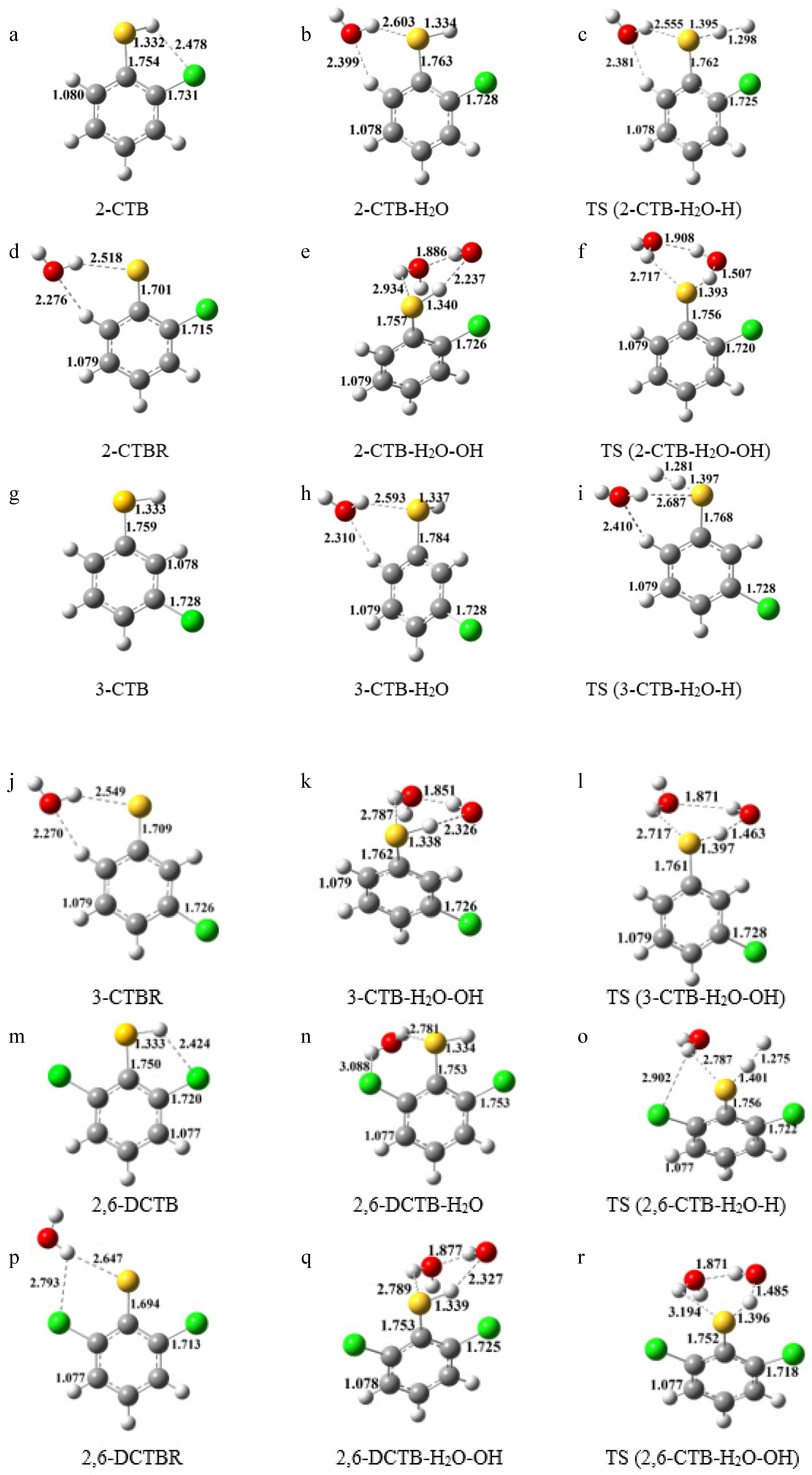
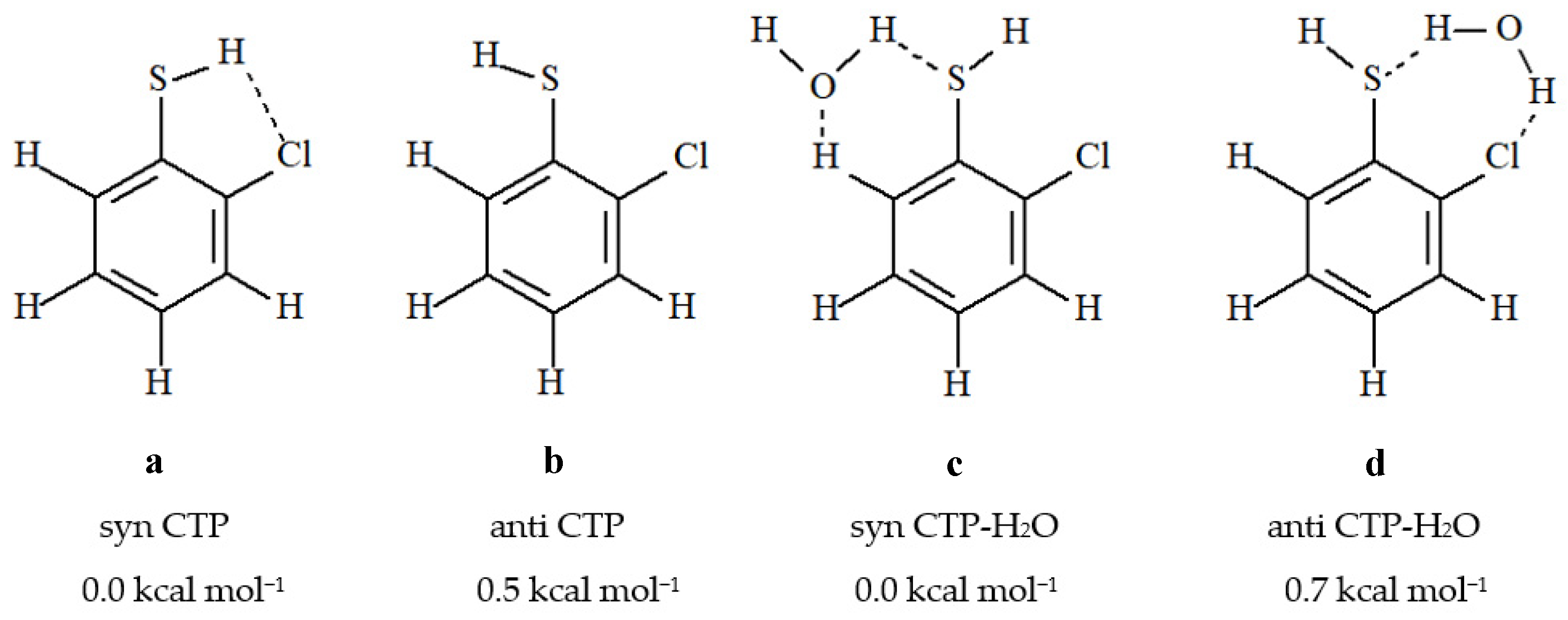
2.1. Effect of Water on the Structural Parameters of Chlorothiobenzene Molecules
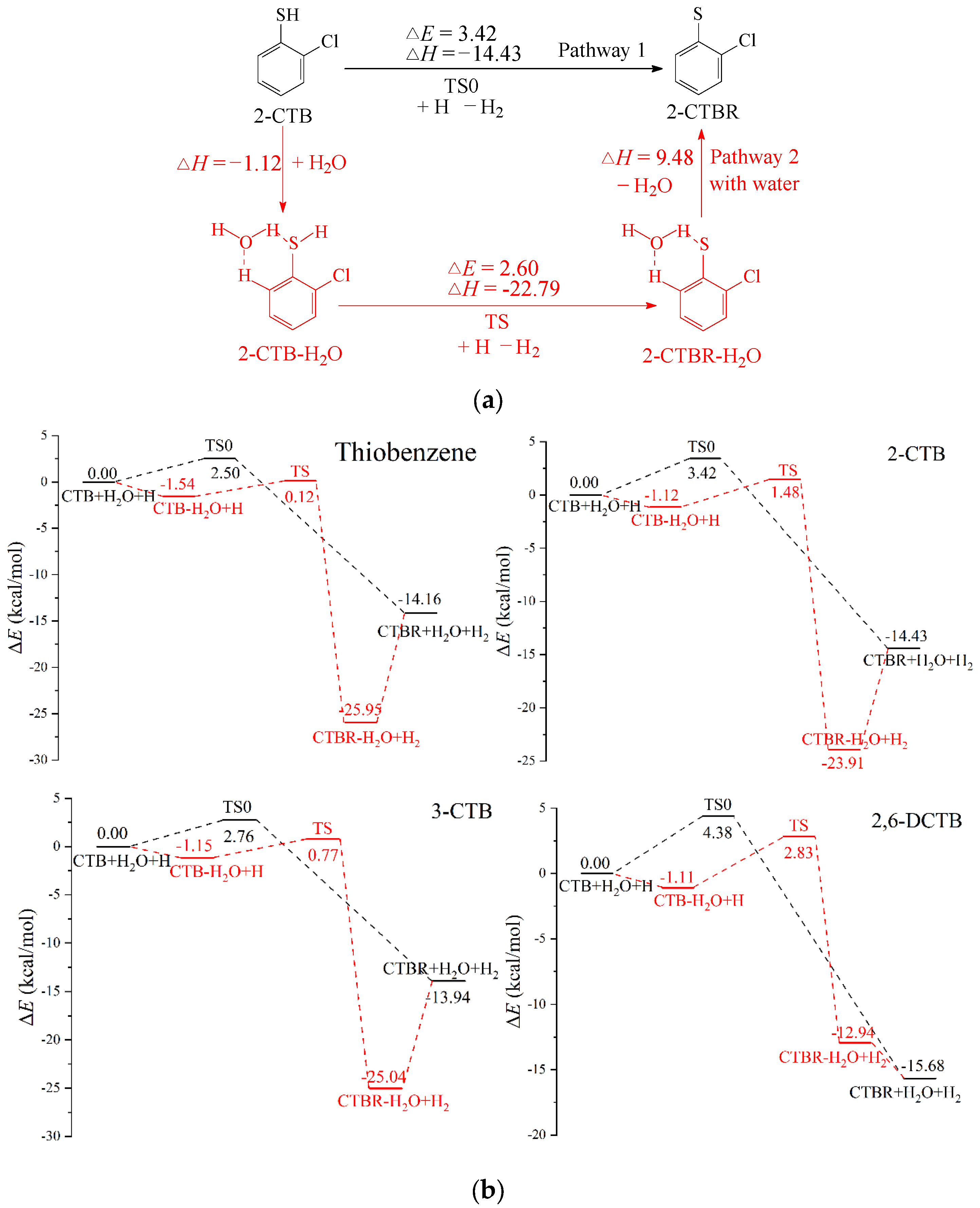
2.2. H Abstraction from CTB-H2Os by H Atom
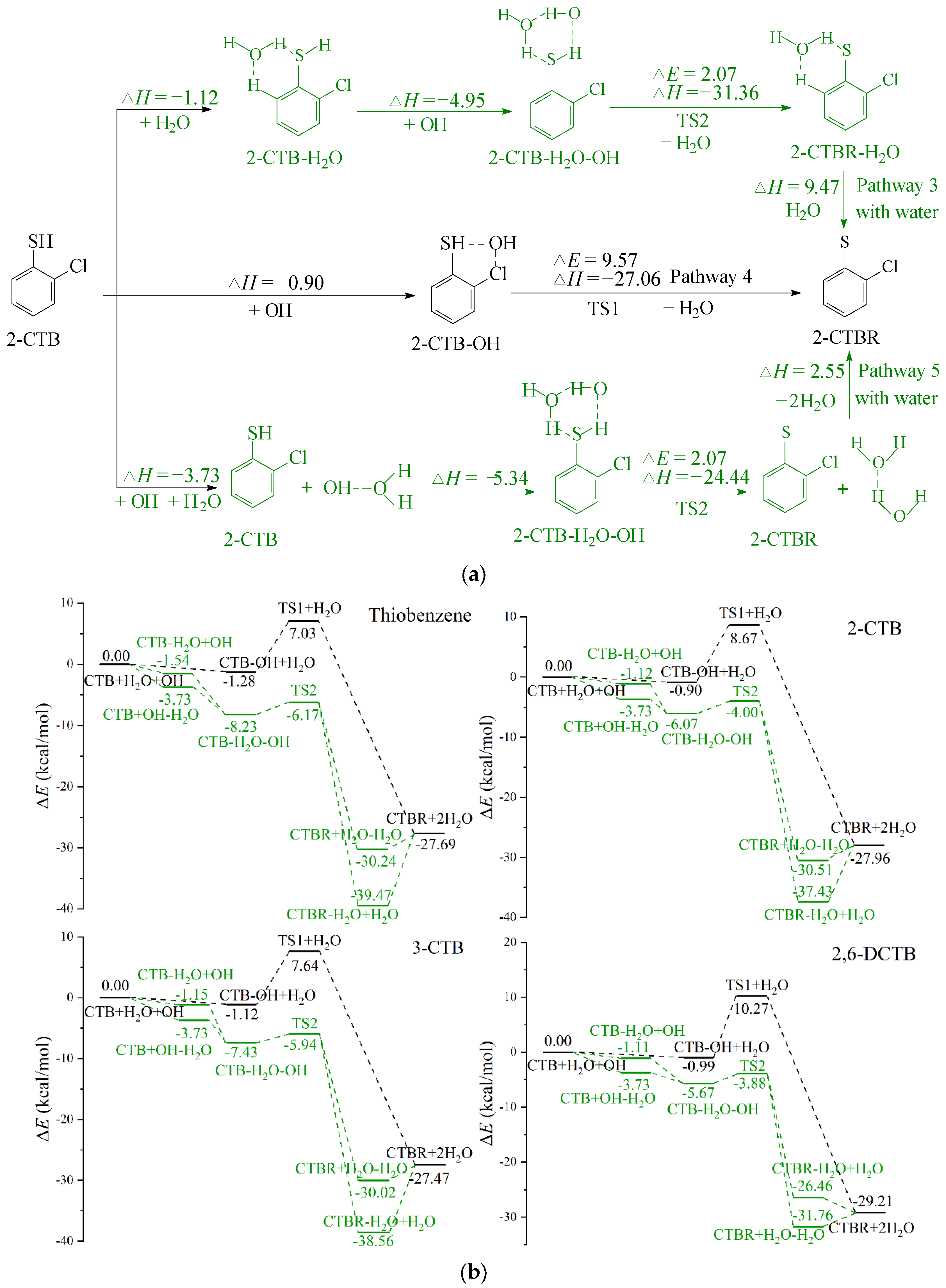
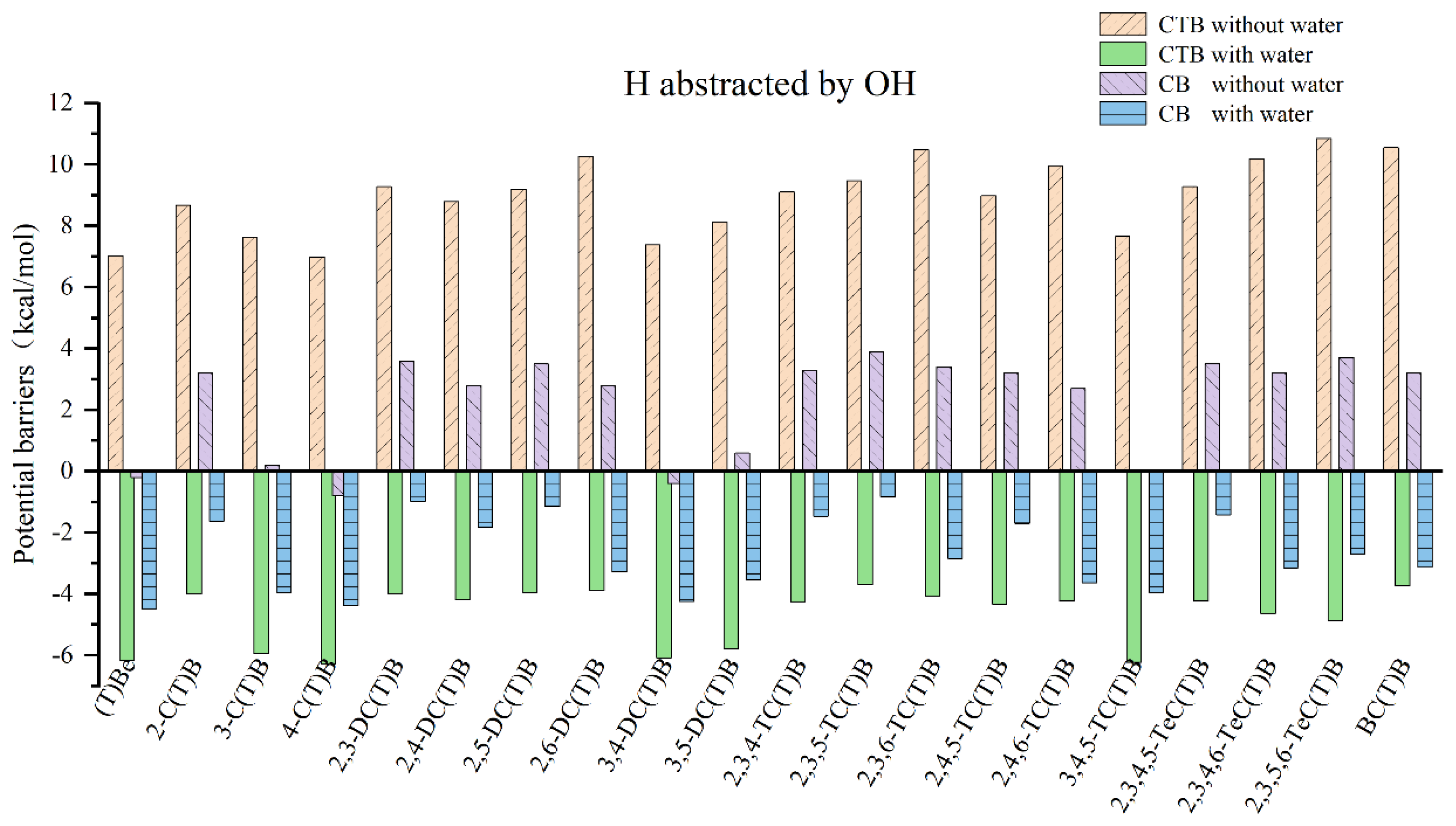
2.3. H Abstraction from CTB-H2Os by ·OH Radicals
2.4. Comparison of H Abstraction from CTB-H2Os by H and ·OH
2.5. Comparison of H Abstraction from CB-H2Os and CTB-H2Os by H/·OH
3. Materials and Methods
3.1. Density Functional Theory
3.2. Accuracy Verification
4. Conclusions
- The presence of water appears to weaken the reactivity of the S–H bonds in CTBs and stabilize the CTB-H2O-H and CTB-H2O-OH transition states, resulting from the existence of the hydrogen bonds in the hydrated CTBs and transition states. Therefore, the introduction of the water molecule can promote the formation of CTBRs from the chlorothiobenzyl-hydrogen abstraction reactions of CTBs with H/·OH.
- The structural parameters and thermal data as well as the formation potential of CTBRs from CTBs by H and ·OH are strongly dominated by the chlorine substitution at the ortho-position of CTBs, but not by the number of the chlorine substituents.
- The water-assistant H abstraction from CTBs by ·OH occurs more easily than the chlorothiobenzyl-hydrogen abstraction by H; the water molecule has a more effective catalytic effect on the H abstraction from CTBs by ·OH than from CTBs by H.
- The water-assistant H abstractions from CTBs by H/·OH are more likely to occur than those from CBs by H/·OH; the water molecule can play a considerably more positive catalytic role in the H abstraction from CTBs by H/·OH than from CBs by H/·OH.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Zeng, X.L.; Chen, H.J.; Wang, H.J. Thermodynamic properties and relative stability of polychlorinated thianthrenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1442–1448. [Google Scholar] [CrossRef]
- Chen, S.D.; Liu, H.X.; Wang, Z.Y. Study of structural and thermodynamic properties for polychlorinated dibenzothiophenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1195–1202. [Google Scholar] [CrossRef]
- Sinkkonen, S. New Types of Persistent Halogenated Compounds; Springer: Berlin/Heildeberg, Germany, 2000; Volume 3, pp. 289–314. [Google Scholar]
- Puzyn, T.; Rostkowski, P.; Świeczkowski, A.; Jędrusiak, A.; Falandysz, J. Prediction of environmental partition coefficients and the Henry’s law constants for 135 congeners of chlorodibenzothiophene. Chemosphere 2006, 62, 1817–1828. [Google Scholar] [CrossRef]
- Kopponen, P.; Sinkkonen, S.; Poso, A.; Gynther, J.; Karenlampi, S. Sulfur analogues of polychlorinated dibenzo-P-dioxins, dibenzofurans and diphenyl ethers as inducers of CYP1A1 in mouse hepatoma cell culture and structure-activity relationships. Environ. Toxicol. Chem. Int. J. 1994, 13, 1543–1548. [Google Scholar] [CrossRef]
- Weber, R.; Hagenmaier, H.; Schrenk, D. Elimination kinetics and toxicity of 2,3,7,8-tetrachlorothiantheren, a thio analogue of 2,3,7,8-TCDD. Chemosphere 1998, 36, 2635–2641. [Google Scholar] [CrossRef]
- Nakai, S.; Kishita, S.; Nomura, Y.; Hosomi, M. Polychlorinated dibenzothiophenes in Japanese environmental samples and their photodegradability and dioxin-like endocrine-disruption potential. Chemosphere 2007, 67, 1852–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, S.; Espino, M.P.; Nomura, Y.; Hosomi, M. Detection of polychlorinated dibenzothiophenes (PCDTs) in the environmental samples and investigation of their photodegradability and dioxin-like endocrine disruption potentcy. J. Environ. Chem. Eng. 2004, 14, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Sinkkonen, S.; Paasivirta, J.; Lahtiperä, M. Chlorinated and methylated dibenzothiophenes in sediment samples from a river contaminated by organochlorine wastes. J. Soil. Sediment. 2001, 1, 9–14. [Google Scholar] [CrossRef]
- Sinkkonen, S. Sources and environmental fate of PCDTs. Toxicol. Environ. Chem. 1998, 66, 105–112. [Google Scholar] [CrossRef]
- Sinkkonen, S. PCDTs in the environment. Chemosphere 1997, 34, 2585–2594. [Google Scholar] [CrossRef]
- Menon, S.; Hansen, J.; Nazarenko, L.; Luo, Y. Climate effects of black carbon aerosols in China and India. Science 2002, 297, 2250–2253. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Chen, Y.; Zhao, Z.; Li, Q.; Zhou, Q.; Ye, Z.; Ge, X. Characteristics, sources and health risks of toxic species (PCDD/Fs, PAHs and heavy metals) in PM2.5 during fall and winter in an industrial area. Chemosphere 2020, 238, 124620. [Google Scholar] [CrossRef]
- Yu, F.; Cui, K.; Sheu, H.-L.; Hsieh, Y.-K.; Tian, X. Atmospheric concentration, particle-bound content, and dry deposition of PCDD/FS. Aerosol. Air Qual. Res. 2021, 21, 210059. [Google Scholar] [CrossRef]
- Koester, C.J.; Hites, R.A. Photodegradation of polychlorinated dioxins and dibenzofurans adsorbed to fly ash. Environ. Sci. Technol. 1992, 26, 502–507. [Google Scholar] [CrossRef]
- Lohmann, R.; Gioia, R.; Eisenreich, S.J.; Jones, K.C. Assessing the importance of ab-and adsorption to the gas-particle partitioning of PCDD/Fs. Atmos. Environ. 2007, 41, 7767–7777. [Google Scholar] [CrossRef]
- Buser, H.R. Identification and sources of dioxin-like compounds: I. Polychlorodibenzothiophenes and polychlorothianthrenes, the sulfur-analogues of the polychlorodibenzofurans and polychlorodibenzodioxins. Chemosphere 1992, 25, 45–48. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Paasivirta, J.; Koistinen, J.; Tarhanen, J. Tetra-and pentachlorodibenzothiophenes are formed in waste combustion. Chemosphere 1991, 23, 583–587. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Kolehmainen, E.; Koistinen, J.; Lahtiperä, M. High-resolution gas chromatographic-mass spectrometric determination of neutral chlorinated aromatic sulphur compounds in stack gas samples. J. Chromatogr. A 1993, 641, 309–317. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Vattulainen, A.; Aittola, J.P.; Paasivirta, J.; Tarhanen, J.; Lahtiperä, M. Metal reclamation produces sulphur analogues of toxic dioxins and furans. Chemosphere 1994, 28, 1279–1288. [Google Scholar] [CrossRef]
- Czerwiński, J. Pathways of polychlorinated dibenzothiophenes (PCDTs) in the environment. Archi. Environ. Prot. 2008, 34, 169–181. [Google Scholar]
- Parette, R.; Pearson, W.N. 2,4,6,8-Tetrachlorodibenzothiophene in the Newark Bay Estuary: The likely source and reaction pathways. Chemosphere 2014, 111, 157–163. [Google Scholar] [CrossRef]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Theoretical study in the dimerisation of 2-chlrothiophenol/2-chlorothiobenzyl: Precursors to PCDT/TA. Organohalog. Compd. 2012, 74, 657–660. [Google Scholar]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Quantum chemical study on formation of PCDT/TA from 2-chlorothiophenol precursor. Environ. Sci. Technol. 2013, 47, 11040–11047. [Google Scholar] [CrossRef]
- Navarro, R.; Bierbrauer, K.; Mijangos, C.; Goiti, E.; Reinecke, H. Modification of poly(vinyl chloride) with new aromatic thiol compounds. Synthesis and characterization. Polym. Degrad. Stab. 2008, 93, 585–591. [Google Scholar] [CrossRef]
- Benz, T.; Hagenmaier, H.; Lindig, C.; She, J. Occurrence of the sulphur analogue of octachlorodibenzo-p-dioxin in the environment and investigations on its potential source. Fresen. J. Anal. Chem. 1992, 344, 286–291. [Google Scholar] [CrossRef]
- Yu, X.Q.; Chang, J.M.; Liu, X.; Pan, W.X.; Zhang, A.Q. Theoretical study on the formation mechanism of polychlorinated dibenzothiophenes/thianthrenes from 2-chlorothiophenol molecules. J. Environ. Sci. 2018, 66, 318–327. [Google Scholar] [CrossRef]
- Wang, H.; Zuo, C.; Zheng, S.; Sun, Y.; Xu, F.; Zhang, Q. Mechanistic and Kinetic Study on Self-/Cross-Condensation of PCTA/DT Formation Mechanisms from Three Types of Radicals of 2,4-Dichlorothiophenol. Int. J. Mol. Sci. 2019, 20, 2623. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Shi, X.; Li, Y.; Zhang, Q. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCTA/DTs from 2,4-dichlorothiophenol and 2,4,6-trichlorothiophenol. Int. J. Mol. Sci. 2015, 16, 20449–20467. [Google Scholar] [CrossRef] [Green Version]
- Zuo, C.; Wang, H.; Pan, W.; Zheng, S.; Xu, F.; Zhang, Q. Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors. Int. J. Mol. Sci. 2019, 20, 1542. [Google Scholar] [CrossRef] [Green Version]
- Shao, K.; Yan, J.; Li, X.; Lu, S.; Wei, Y.; Fu, M. Experimental study on the effects of H2O on PCDD/Fs formation by de novo synthesis in carbon/CuCl2 model system. Chemosphere 2010, 78, 672–679. [Google Scholar] [CrossRef]
- Li, X.-D.; Zhang, J.; Yan, J.-H.; Chen, T.; Lu, S.-Y.; Cen, K.-F. Effect of water on catalyzed de novo formation of polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans. J. Hazard. Mater. 2006, 137, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Briois, C.; Ryan, S.; Tabor, D.; Touati, A.; Gullett, B.K. Formation of polychlorinated dibenzo-p-dioxins and dibenzofurans from a mixture of chlorophenols over fly ash: Influence of water vapor. Environ. Sci. Technol. 2007, 41, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, L.; Zwick, G.; Beck, J.; Bautz, H.; Roth, W.J.C. The role of particulate carbon in the de-novo synthesis of polychlorinated dibenzodioxins and-furans in fly-ash. Chemosphere 1990, 20, 1953–1958. [Google Scholar] [CrossRef]
- Jay, K.; Stieglitz, L. On the mechanism of formation of polychlorinated aromatic-compounds with copper(II) chloride. Chemosphere 1991, 22, 987–996. [Google Scholar] [CrossRef]
- Shi, X.L.; Zhang, R.M.; Zhang, H.J.; Xu, F.; Zhang, Q.Z.; Wang, W.X. Influence of water on the homogeneous gas-phase formation mechanism of polyhalogenated dioxins/furans from chlorinated/brominated phenols as precursors. Chemosphere 2015, 137, 142–148. [Google Scholar] [CrossRef]
- Xu, F.; Shi, X.L.; Zhang, Q.Z.; Wang, W.X. Formation of Chlorotriophenoxy Radicals from Complete Series Reactions of Chlorotriophenols with H and OH Radicals. Int. J. Mol. Sci. 2015, 16, 18714–18731. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Z.; Qu, X.H.; Wang, H.T.; Xu, F.; Shi, X.L.; Wang, W.X. Mechanism and Thermal Rate Constants for the Complete Series Reactions of Chlorophenols with H. Environ. Sci. Technol. 2009, 43, 4105–4112. [Google Scholar] [CrossRef]
- Xu, F.; Wang, H.T.; Zhang, Q.; Zhang, R.M.; Qu, X.H.; Wang, W.X. Kinetic properties for the complete series reactions of chlorophenols with OH radicals-relevance for dioxin formation. Environ. Sci. Technol. 2010, 44, 1399–1404. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTBs | L(S−H) | CTBRs | L(S−C) | D0(S−H) a | D0(S−H) b | ||
|---|---|---|---|---|---|---|---|
| Non-Hydrated a | Hydrated b | Non-Hydrated a | Hydrated b | ||||
| TBe | 1.33 | 1.34 | TbeR | 1.76 | 1.71 | 86.51 | 76.26 |
| 2-CTB | 1.33 | 1.33 | 2-CTBR | 1.74 | 1.70 | 86.24 | 77.87 |
| 3-CTB | 1.33 | 1.34 | 3-CTBR | 1.75 | 1.71 | 86.73 | 76.77 |
| 4-CTB | 1.33 | 1.34 | 4-CTBR | 1.71 | 1.70 | 77.16 | 75.59 |
| 2,3-DCTB | 1.33 | 1.33 | 2,3-DCTBR | 1.74 | 1.70 | 85.59 | 78.45 |
| 2,4-DCTB | 1.33 | 1.33 | 2,4-DCTBR | 1.70 | 1.70 | 79.15 | 77.32 |
| 2,5-DCTB | 1.33 | 1.33 | 2,5-DCTBR | 1.74 | 1.70 | 87.76 | 78.91 |
| 2,6-DCTB | 1.33 | 1.33 | 2,6-DCTBR | 1.74 | 1.69 | 84.99 | 88.83 |
| 3,4-DCTB | 1.33 | 1.34 | 3,4-DCTBR | 1.71 | 1.71 | 77.98 | 76.26 |
| 3,5-DCTB | 1.33 | 1.33 | 3,5-DCTBR | 1.74 | 1.71 | 86.92 | 77.56 |
| 2,3,4-TCTB | 1.33 | 1.33 | 2,3,4-TCTBR | 1.70 | 1.70 | 79.57 | 77.75 |
| 2,3,5-TCTB | 1.33 | 1.33 | 2,3,5-TCTBR | 1.74 | 1.70 | 86.01 | 79.23 |
| 2,3,6-TCTB | 1.33 | 1.33 | 2,3,6-TCTBR | 1.74 | 1.70 | 87.64 | 88.62 |
| 2,4,5-TCTB | 1.33 | 1.33 | 2,4,5-TCTBR | 1.70 | 1.70 | 79.72 | 77.91 |
| 2,4,6-TCTB | 1.33 | 1.33 | 2,4,6-TCTBR | 1.69 | 1.69 | 80.55 | 80.02 |
| 3,4,5-TCTB | 1.33 | 1.33 | 3,4,5-TCTBR | 1.71 | 1.71 | 78.55 | 76.97 |
| 2,3,4,5-TeCTB | 1.33 | 1.33 | 2,3,4,5-TeCTBR | 1.70 | 1.70 | 80.10 | 78.62 |
| 2,3,4,6-TeCTB | 1.33 | 1.33 | 2,3,4,6-TeCTBR | 1.69 | 1.69 | 81.02 | 79.75 |
| 2,3,5,6-TeCTB | 1.33 | 1.33 | 2,3,5,6-TeCTBR | 1.74 | 1.70 | 84.18 | 88.11 |
| PCTB | 1.33 | 1.33 | PCTBR | 1.70 | 1.69 | 81.69 | 80.82 |
| CTBs | Non-Hydrated a | Hydrated b | ΔE−ΔEH2O | Imaginary Frequencies | ||
|---|---|---|---|---|---|---|
| ΔE | ΔH | ΔEH2O | ΔHH2O | |||
| TBe | 2.50 | −14.16 | 0.12 | −14.16 | 2.38 | −794i |
| 2-CTB | 3.42 | −14.43 | 1.48 | −14.43 | 1.94 | −868i |
| 3-CTB | 2.76 | −13.94 | 0.77 | −13.94 | 1.99 | −904i |
| 4-CTB | 2.31 | −23.50 | 0.22 | −23.50 | 2.09 | −873i |
| 2,3-DCTB | 3.56 | −14.71 | 1.63 | −14.71 | 1.93 | −883i |
| 2,4-DCTB | 3.44 | −21.52 | 1.37 | −21.52 | 2.07 | −855i |
| 2,5-DCTB | 3.64 | −12.90 | 1.76 | −12.90 | 1.88 | −882i |
| 2,6-DCTB | 4.38 | −15.68 | 2.83 | −15.68 | 1.55 | −922i |
| 3,4-DCTB | 2.65 | −22.68 | 0.92 | −22.68 | 1.73 | −804i |
| 3,5-DCTB | 3.00 | −13.74 | 1.09 | −13.74 | 1.91 | −948i |
| 2,3,4-TCTB | 3.21 | −21.10 | 1.06 | −21.10 | 2.15 | −891i |
| 2,3,5-TCTB | 3.70 | −14.65 | 2.06 | −14.65 | 1.64 | −903i |
| 2,3,6-TCTB | 4.43 | −13.03 | 3.62 | −13.03 | 0.81 | −919i |
| 2,4,5-TCTB | 3.48 | −20.94 | 1.55 | −20.94 | 1.93 | −880i |
| 2,4,6-TCTB | 4.27 | −20.12 | 3.11 | −20.12 | 1.16 | −896i |
| 3,4,5-TCTB | 2.67 | −22.12 | 1.11 | −22.12 | 1.56 | −915i |
| 2,3,4,5-TeCTB | 3.17 | −20.57 | 1.32 | −20.57 | 1.85 | −890i |
| 2,3,4,6-TeCTB | 4.37 | −19.65 | 3.25 | −19.65 | 1.12 | −904i |
| 2,3,5,6-TeCTB | 4.52 | −16.48 | 3.31 | −16.48 | 1.21 | −930i |
| PCTB | 4.62 | −18.98 | 3.29 | −18.98 | 1.33 | −915i |
| CTBs | Non-Hydrated a | Hydrated b | ΔE−ΔEH2O | Imaginary Frequencies | ||
|---|---|---|---|---|---|---|
| ΔE | ΔH | ΔEH2O | ΔHH2O | |||
| TBe | 7.03 | −27.69 | −6.17 | −27.69 | 13.20 | −702i |
| 2-CTB | 8.67 | −27.96 | −4.00 | −27.96 | 12.67 | −643i |
| 3-CTB | 7.64 | −27.47 | −5.94 | −27.47 | 13.58 | −721i |
| 4-CTB | 6.99 | −37.03 | −6.29 | −37.03 | 13.28 | −703i |
| 2,3-DCTB | 9.29 | −28.24 | −4.00 | −28.24 | 13.29 | −714i |
| 2,4-DCTB | 8.80 | −35.05 | −4.19 | −35.05 | 12.99 | −722i |
| 2,5-DCTB | 9.20 | −27.97 | −3.97 | −27.97 | 13.17 | −684i |
| 2,6-DCTB | 10.27 | −29.21 | −3.88 | −29.21 | 14.15 | −783i |
| 3,4-DCTB | 7.39 | −36.21 | −6.08 | −36.21 | 13.47 | −739i |
| 3,5-DCTB | 8.13 | −27.27 | −5.78 | −27.27 | 13.91 | −716i |
| 2,3,4-TCTB | 9.10 | −34.63 | −4.25 | −34.63 | 13.35 | −674i |
| 2,3,5-TCTB | 9.48 | −28.18 | −3.69 | −28.18 | 13.17 | −689i |
| 2,3,6-TCTB | 10.48 | −26.56 | −4.06 | −26.56 | 14.54 | −776i |
| 2,4,5-TCTB | 8.98 | −34.47 | −4.33 | −34.47 | 13.31 | −680i |
| 2,4,6-TCTB | 9.95 | −33.65 | −4.23 | −33.65 | 14.18 | −742i |
| 3,4,5-TCTB | 7.66 | −35.65 | −6.23 | −35.65 | 13.89 | −719i |
| 2,3,4,5-TeCTB | 9.29 | −34.10 | −4.23 | −34.10 | 13.52 | −654i |
| 2,3,4,6-TeCTB | 10.18 | −33.18 | −4.64 | −33.18 | 14.82 | −746i |
| 2,3,5,6-TeCTB | 10.85 | −30.01 | −4.12 | −30.01 | 14.97 | −802i |
| PCTB | 10.55 | −32.51 | −3.73 | −32.51 | 14.28 | −828i |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Zheng, S.; Teng, Z.; Hadizadeh, M.H.; Zhang, Q.; Xu, F.; Sun, Y. Effect of Water Molecule on the Complete Series Reactions of Chlorothiobenzenes with H/·OH: A Theoretical Study. Atmosphere 2022, 13, 849. https://doi.org/10.3390/atmos13050849
Han Y, Zheng S, Teng Z, Hadizadeh MH, Zhang Q, Xu F, Sun Y. Effect of Water Molecule on the Complete Series Reactions of Chlorothiobenzenes with H/·OH: A Theoretical Study. Atmosphere. 2022; 13(5):849. https://doi.org/10.3390/atmos13050849
Chicago/Turabian StyleHan, Yanan, Siyuan Zheng, Zhuochao Teng, Mohammad Hassan Hadizadeh, Qi Zhang, Fei Xu, and Yanhui Sun. 2022. "Effect of Water Molecule on the Complete Series Reactions of Chlorothiobenzenes with H/·OH: A Theoretical Study" Atmosphere 13, no. 5: 849. https://doi.org/10.3390/atmos13050849
APA StyleHan, Y., Zheng, S., Teng, Z., Hadizadeh, M. H., Zhang, Q., Xu, F., & Sun, Y. (2022). Effect of Water Molecule on the Complete Series Reactions of Chlorothiobenzenes with H/·OH: A Theoretical Study. Atmosphere, 13(5), 849. https://doi.org/10.3390/atmos13050849