
Secondary Organic Aerosol (SOA) from Photo-Oxidation of Toluene: 1 Influence of Reactive Nitrogen, Acidity and Water Vapours on Optical Properties


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experiments
2.1.1. Experimental Setup
2.1.2. OH Radical Generation
2.1.3. Experimental Matrix
2.2. Methods
2.2.1. Data Processing
2.2.2. Calculations
2.2.3. Models
3. Results and Discussion
3.1. Summary of Key Experimental Results
3.2. Influence of NOx and RH on SOA (Experiments: Tol-1 to Tol-4)
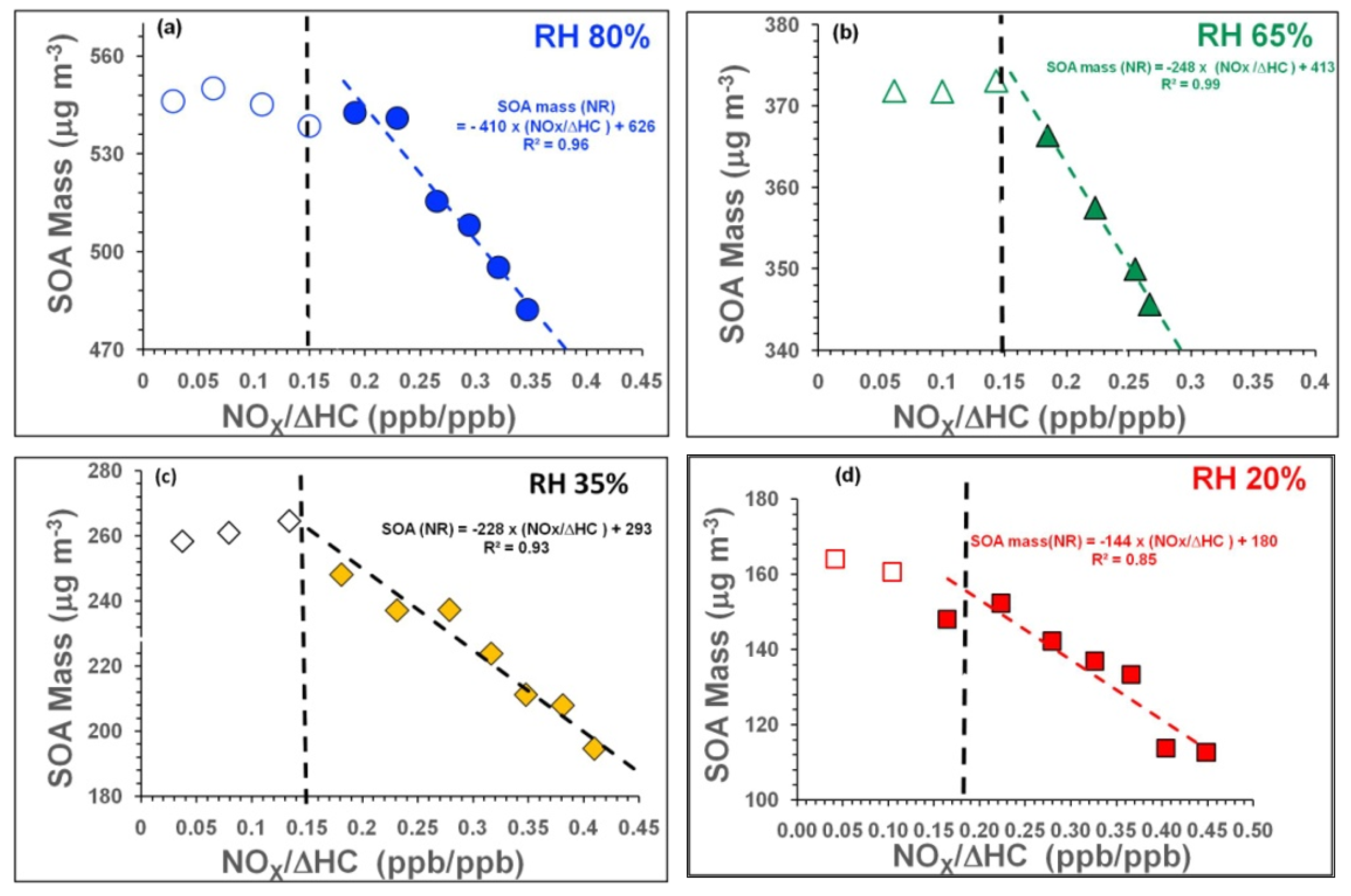
3.2.1. Influence of NOx and RH on SOA Mass Formation
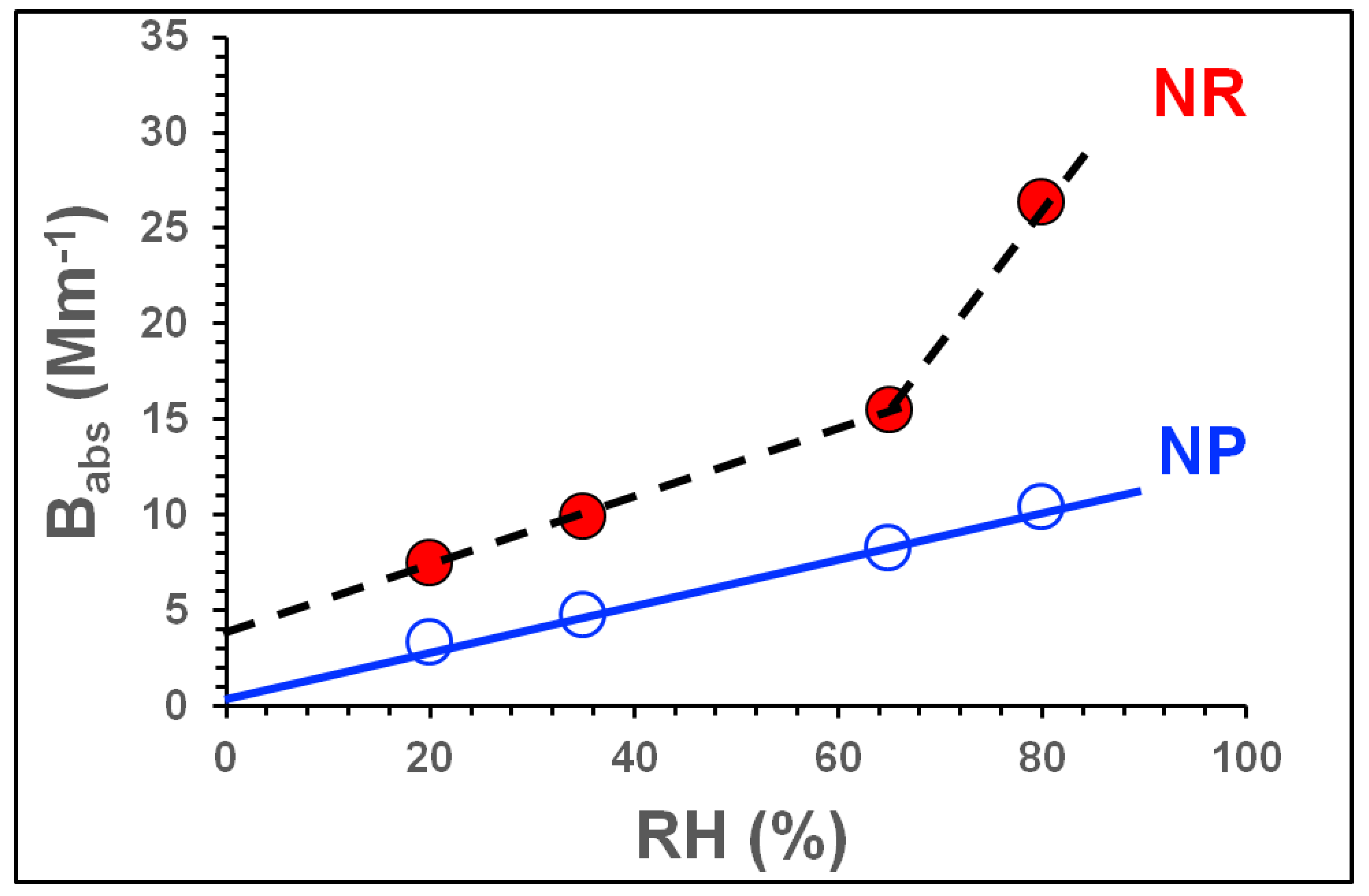
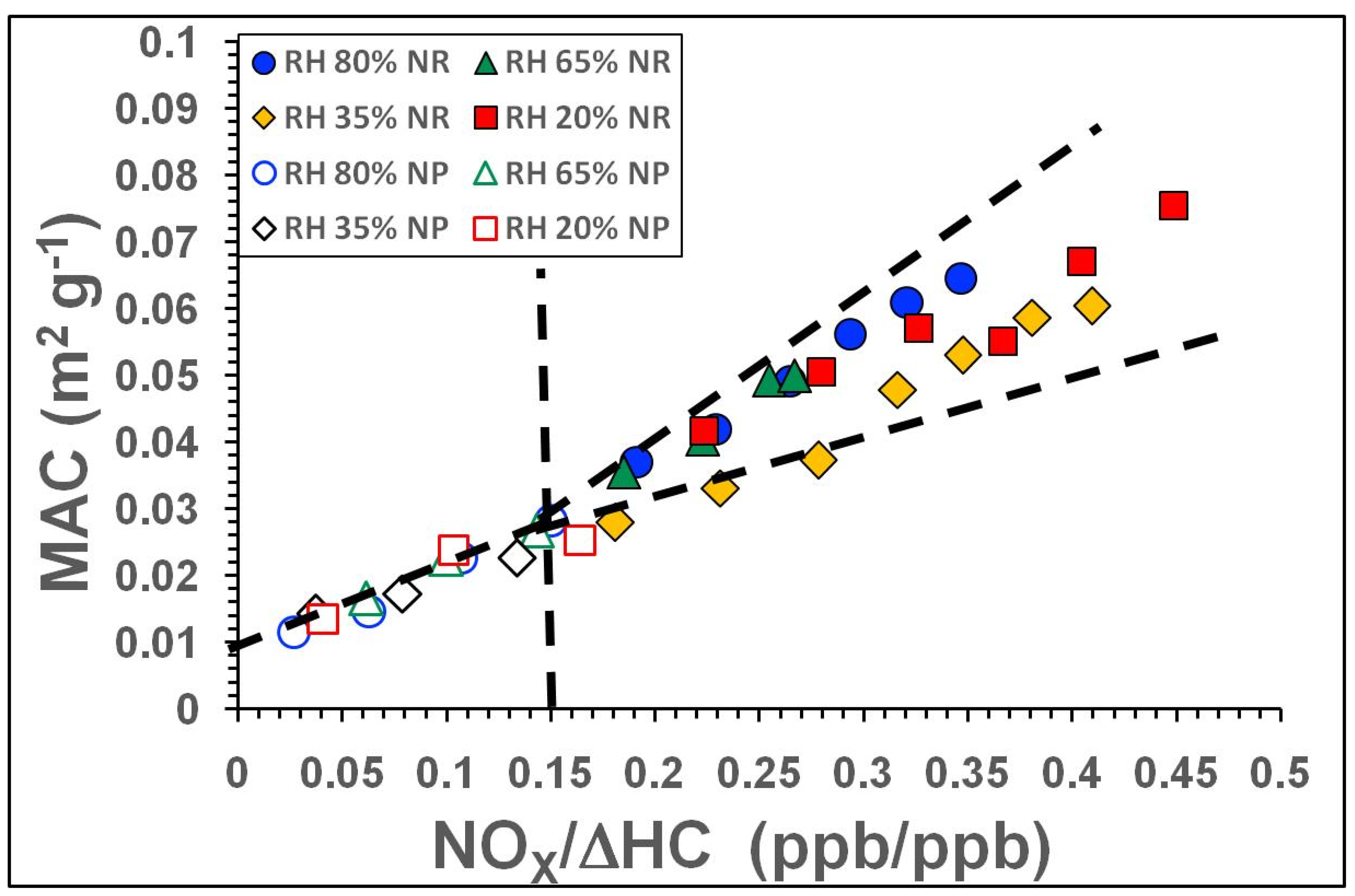
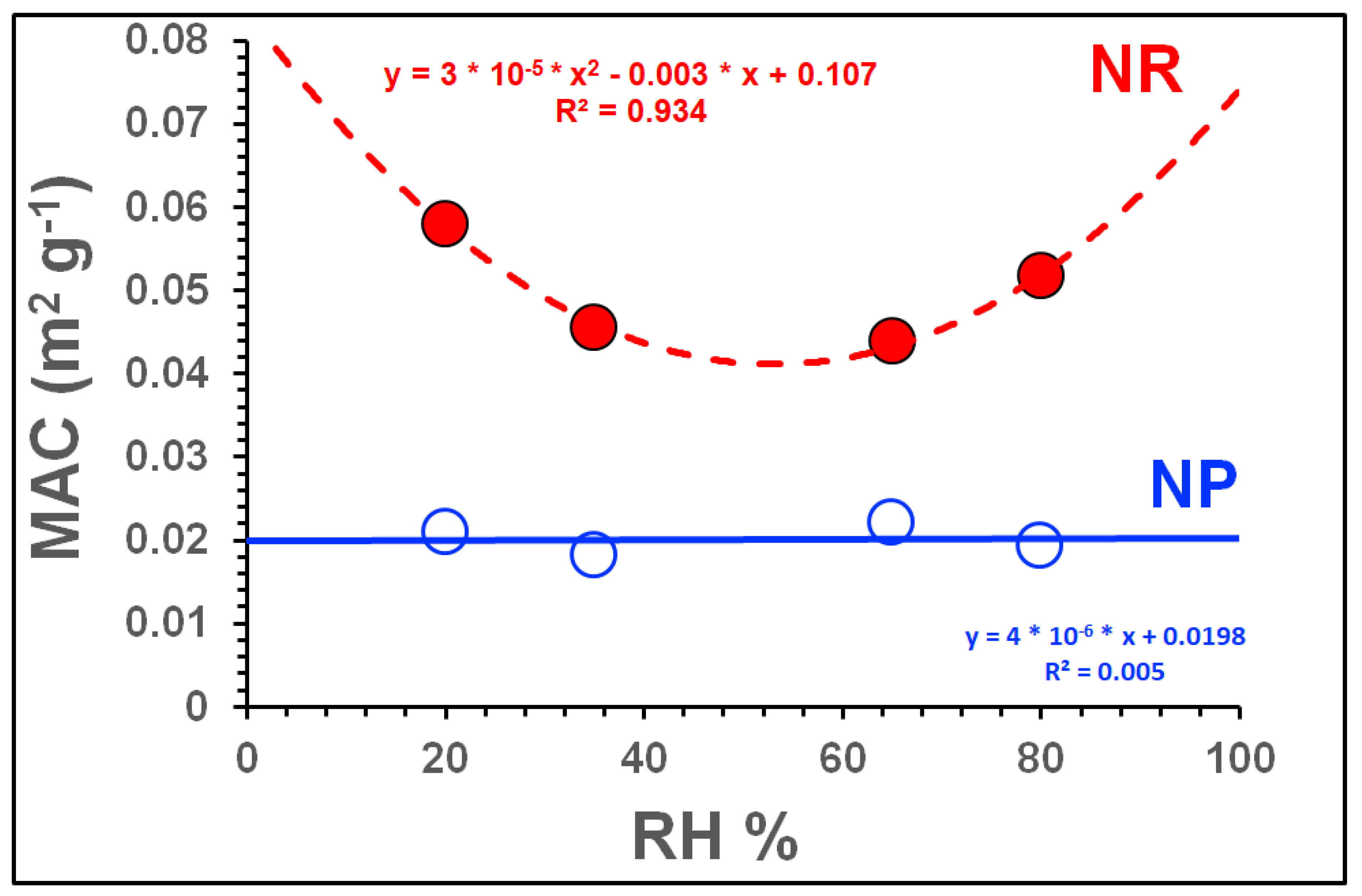
3.2.2. Influence of NOx and RH on Light Absorption
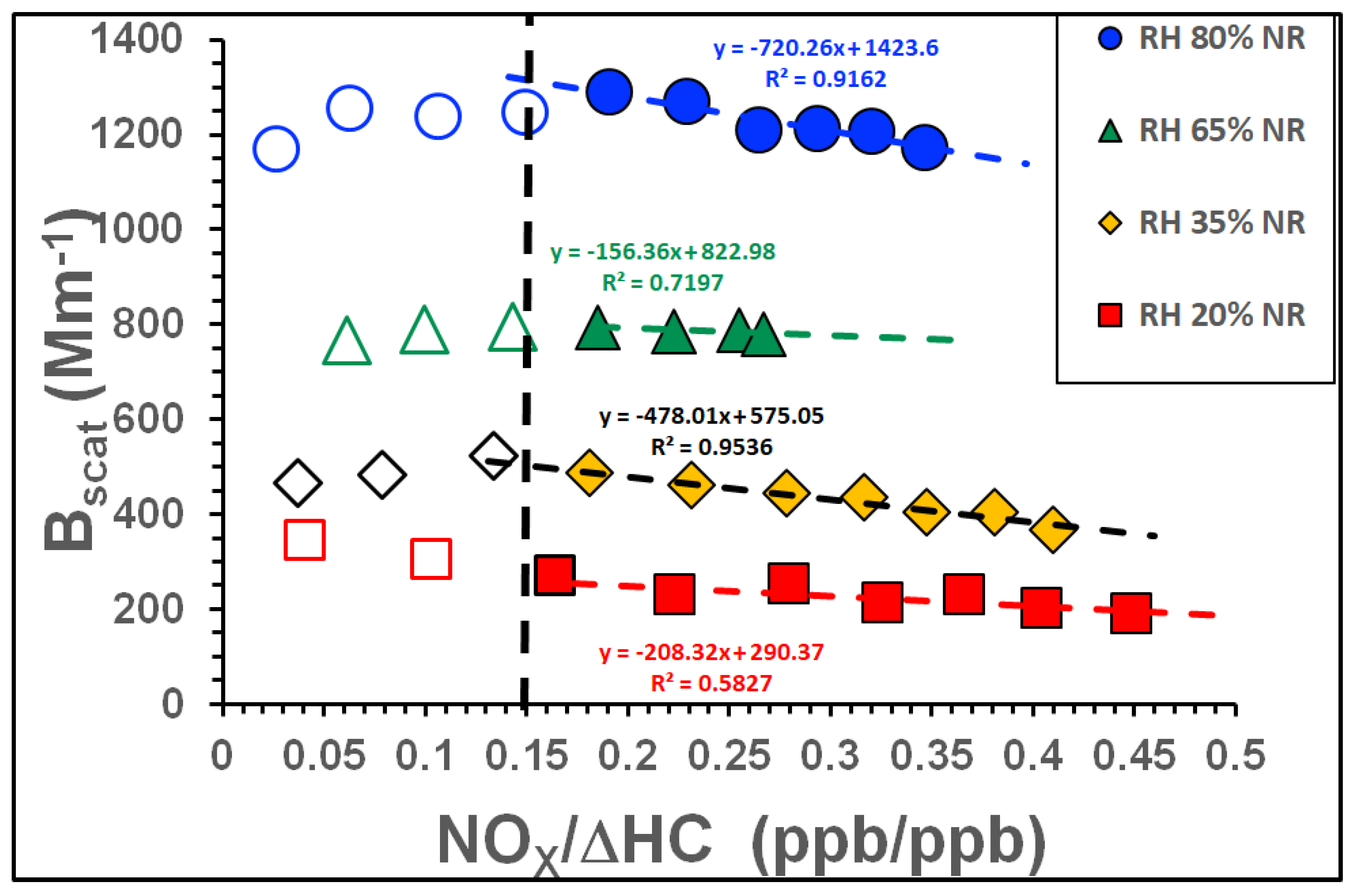
3.2.3. Influence of NOx and RH on Light Scattering
3.3. Influence of NH3 and NOx on SOA at High RH (Experiments: Tol-5 and Tol-6)
3.3.1. Influence of NH3 and NOx on SOA Mass at 80% RH
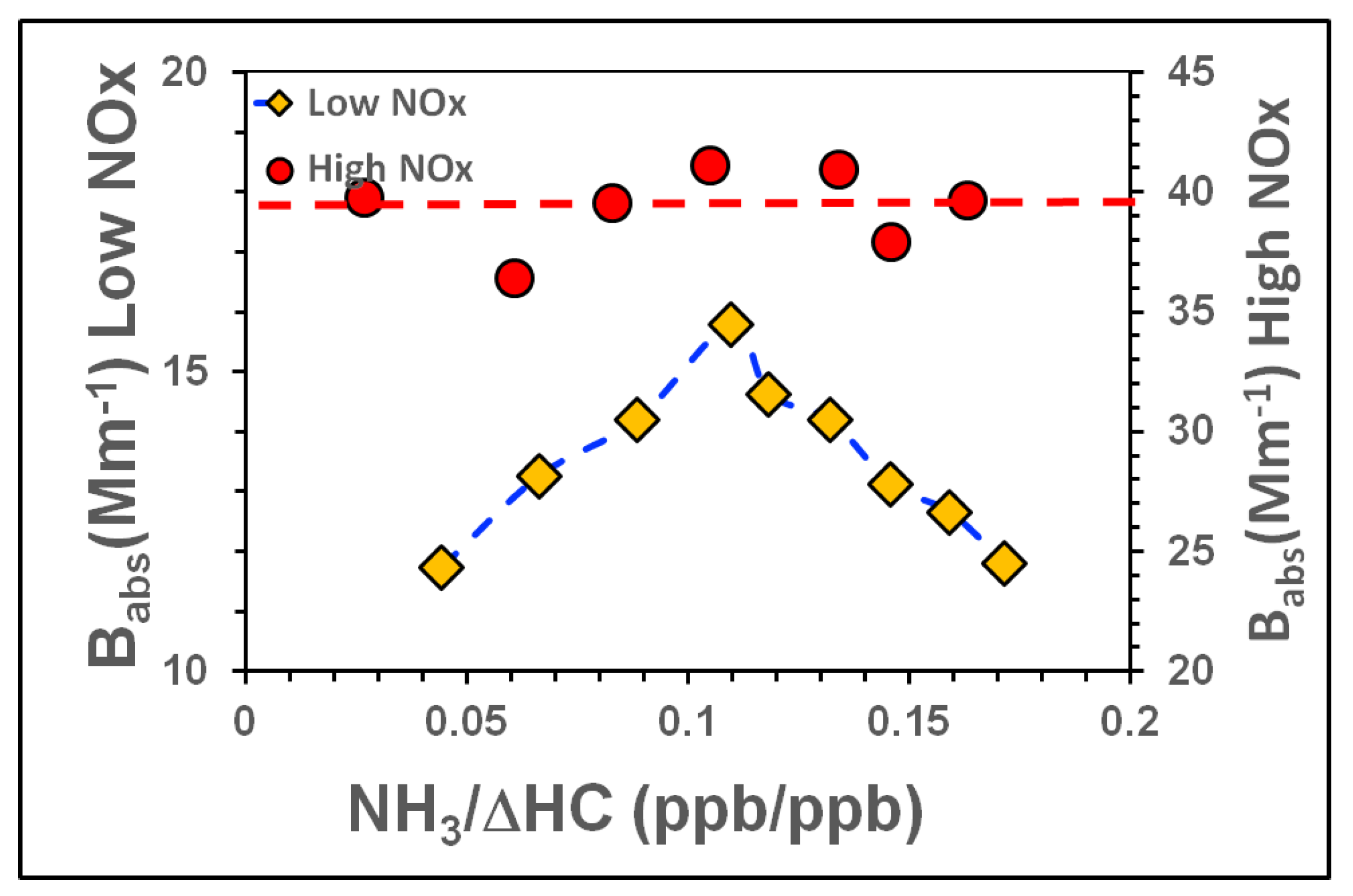
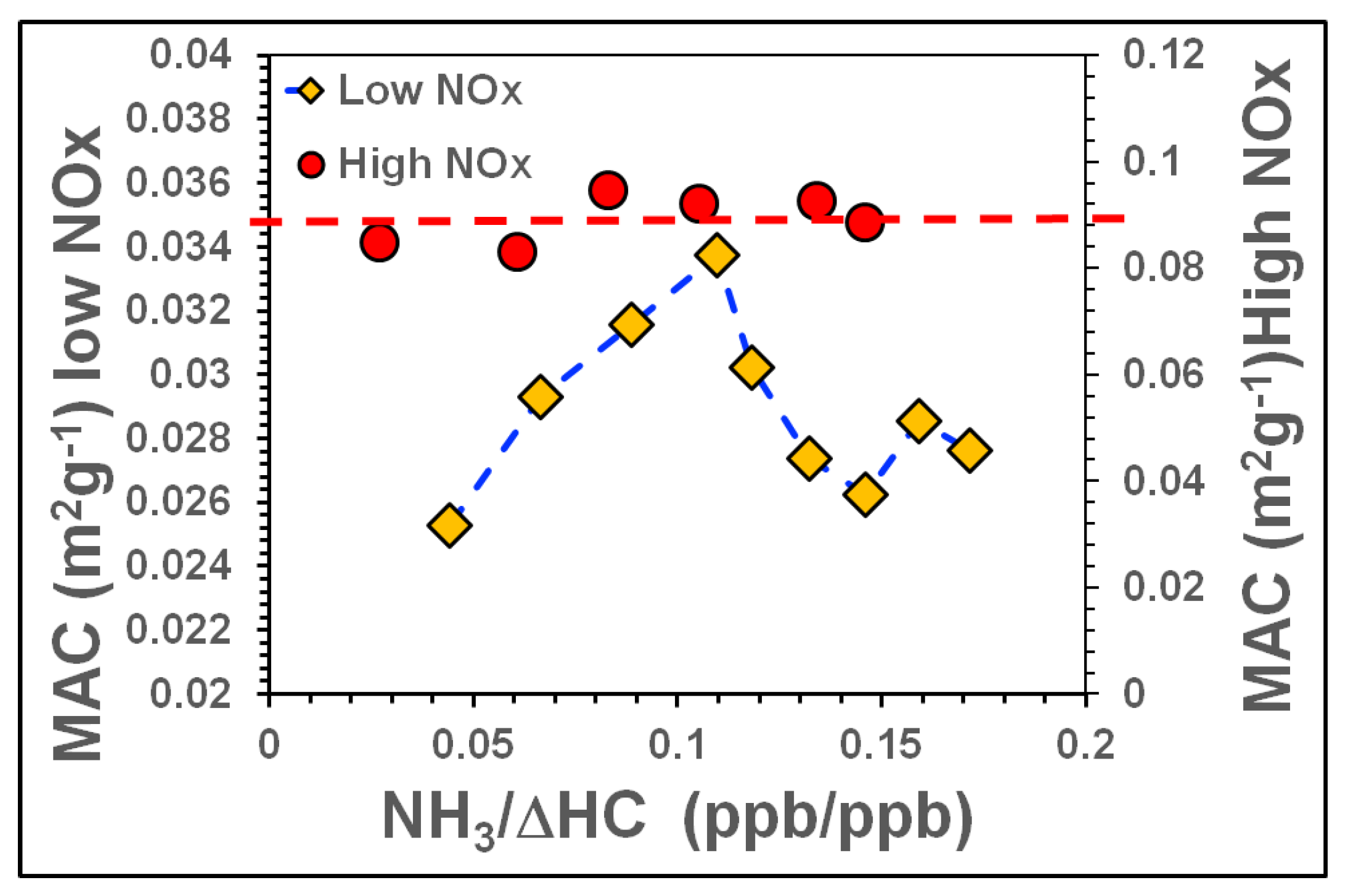
3.3.2. Influence of NH3, NOx on SOA Light Absorption at 80% RH
3.3.3. Influence of NH3 and NOx on SOA Light Scattering at 80% RH
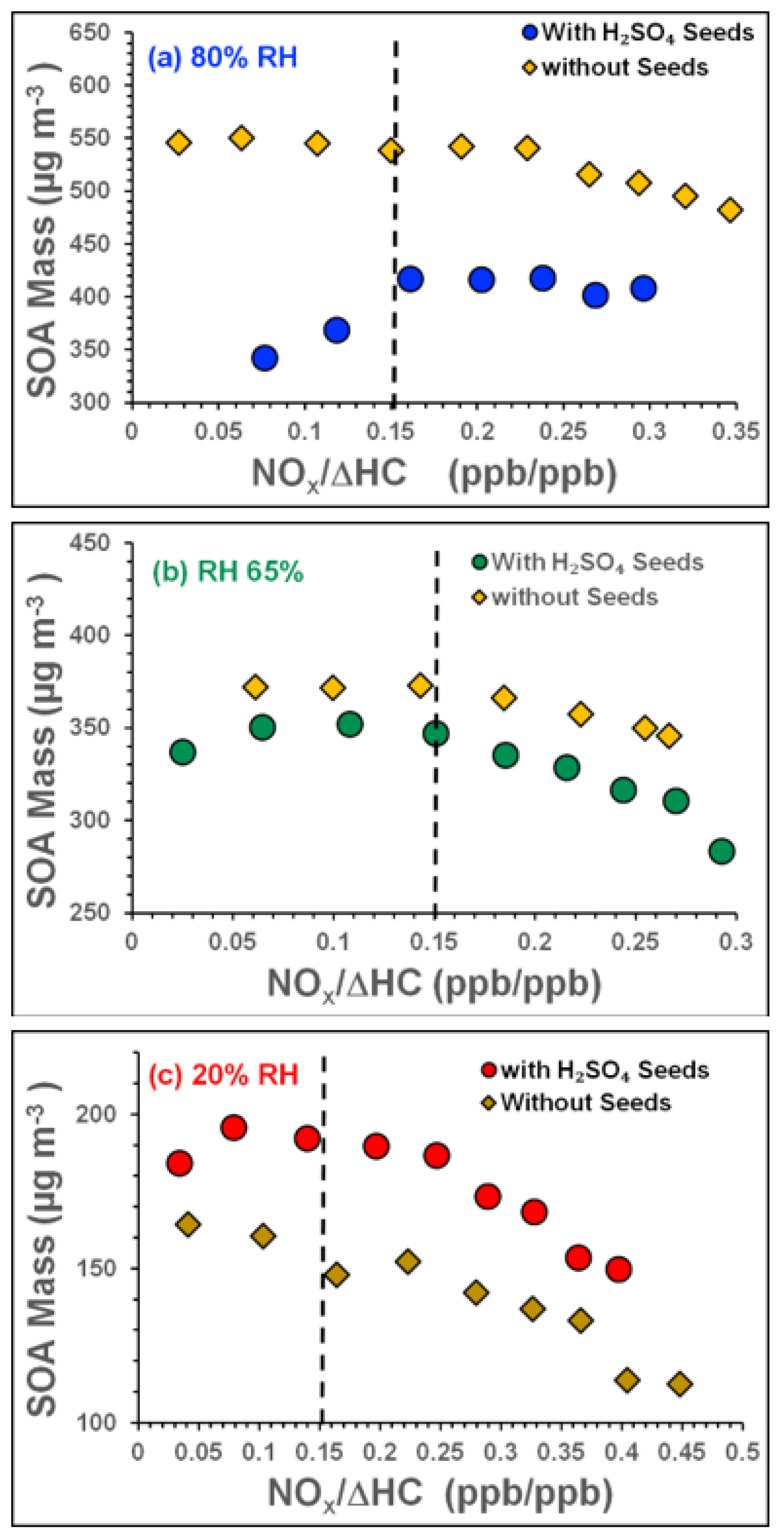
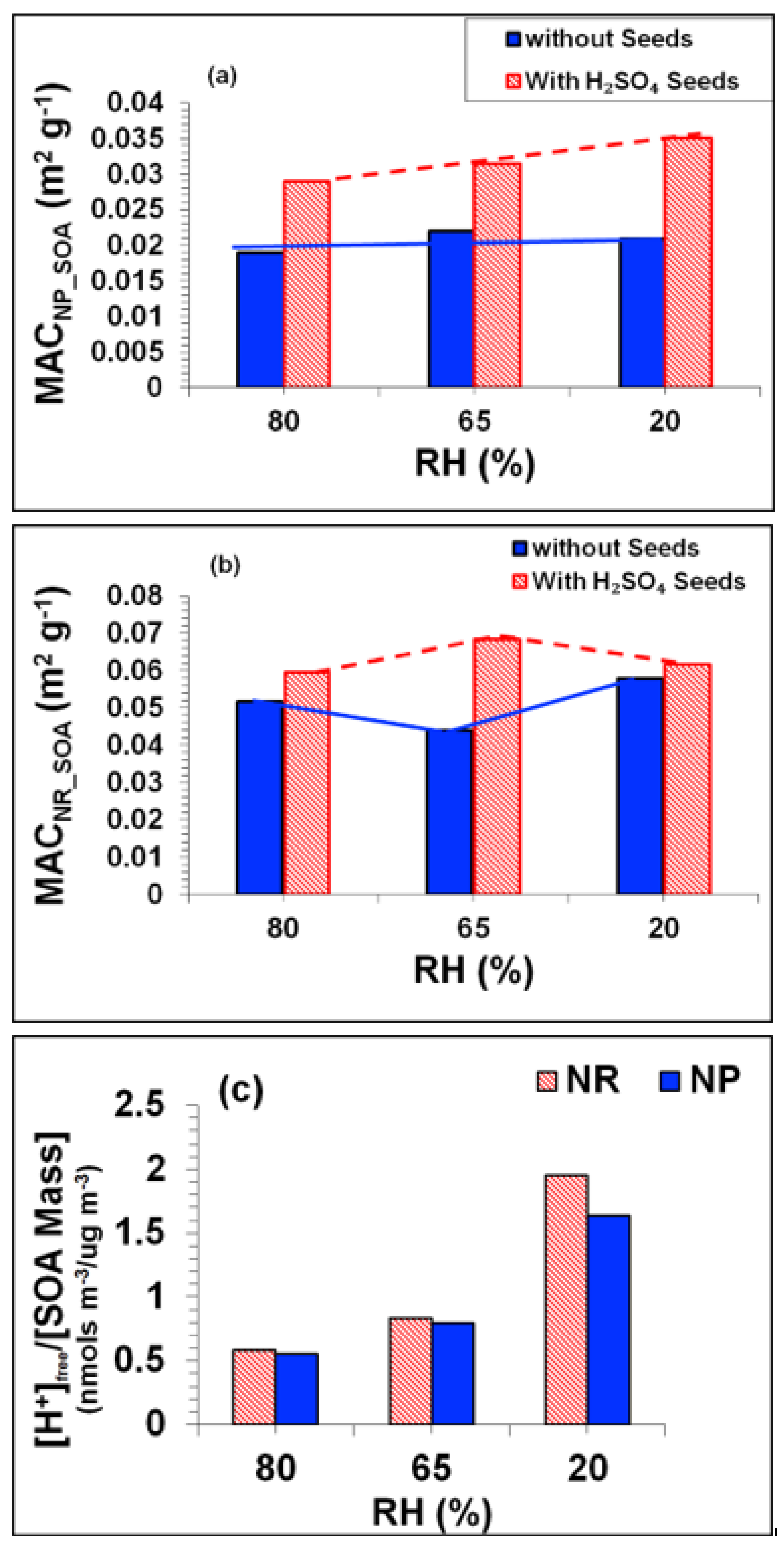
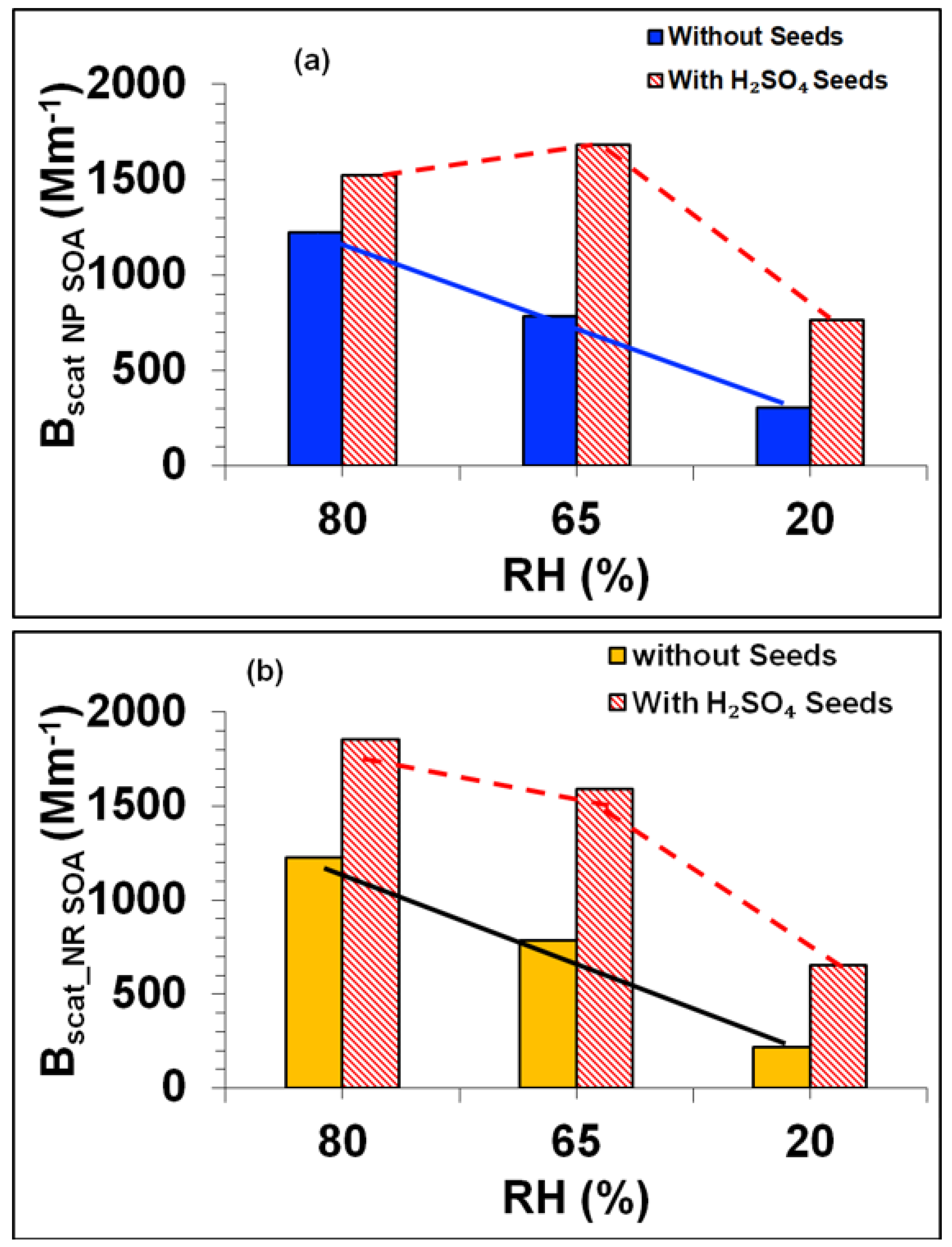
3.4. Influence of NOx Acidity and RH on SOA (Experiments: Tol-7 to Tol-9)
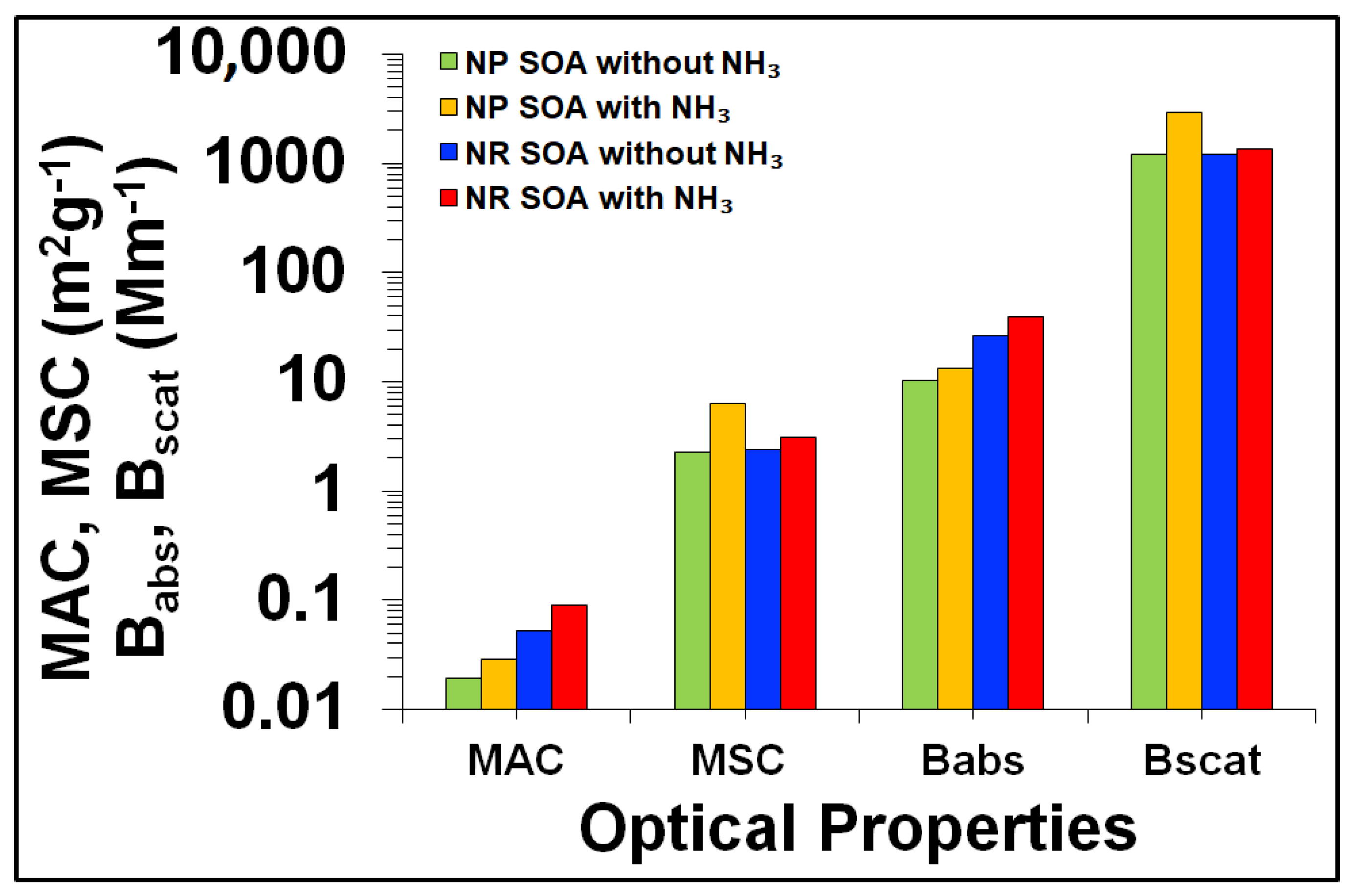
3.4.1. Influence of NOx Acidity and RH on SOA Mass
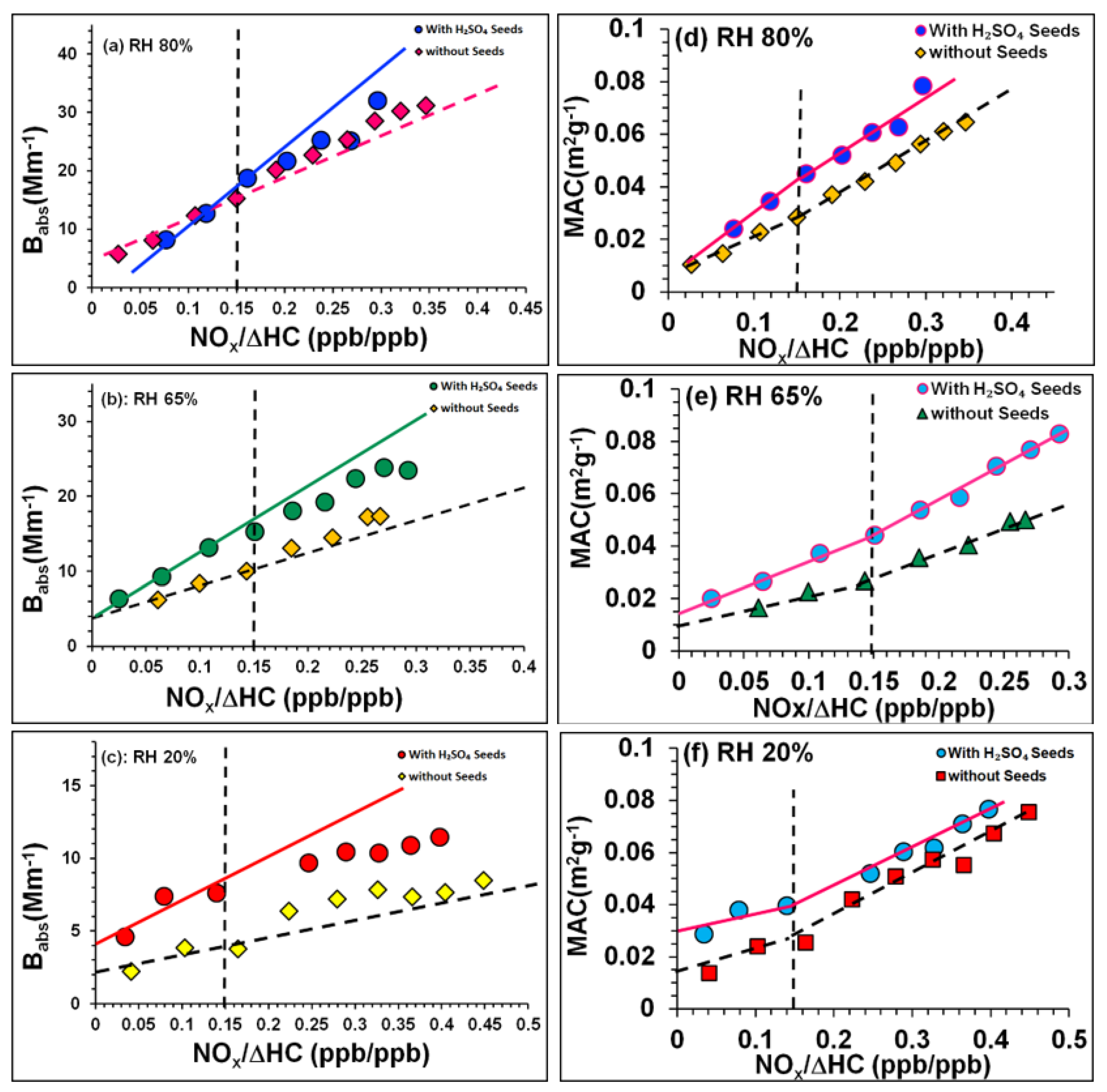
3.4.2. Influence of NOx Acidity and RH on SOA Light Absorption
3.4.3. Influence of NOx Acidity and RH on SOA Light Scattering
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arias, P.A.; Bellouin, N.; Coppola, E.; Jones, R.G.; Krinner, G.; Marotzke, J.; Naik, V.; Palmer, M.D.; Plattner, G.; Rogelj, J.; et al. IPCC, 2021: Technical Summary. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group 15 I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; IPCC AR6 WGI; Cambridge University Press: Cambridge, UK, 2021; pp. 1–3949, in press. [Google Scholar]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef] [Green Version]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Myhre, G.; Samset, B.H.; Schulz, M.; Balkanski, Y.; Bauer, S.; Berntsen, T.K.; Bian, H.; Bellouin, N.; Chin, M.; Diehl, T.; et al. Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations. Atmos. Chem. Phys. 2013, 13, 1853–1877. [Google Scholar] [CrossRef] [Green Version]
- Ervens, B.; Turpin, B.J.; Weber, R.J. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102. [Google Scholar] [CrossRef] [Green Version]
- Herckes, P.; Lee, T.; Trenary, L.; Kang, G.G.; Chang, H.; Collett, J.L. Organic matter in Central California radiation fogs. Environ. Sci. Technol. 2002, 36, 4777–4782. [Google Scholar] [CrossRef]
- Carlton, A.G.; Wiedinmyer, C.; Kroll, J.H. A review of Secondary Organic Aerosol (SOA) formation from isoprene. Atmos. Chem. Phys. 2009, 9, 4987–5005. [Google Scholar] [CrossRef] [Green Version]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Rudich, Y.; Donahue, N.M.; Mentel, T.F. Aging of organic aerosol: Bridging the gap between laboratory and field studies. Annu. Rev. Phys. Chem. 2007, 58, 321–352. [Google Scholar] [CrossRef]
- Kalberer, M.; Paulsen, D.; Sax, M.; Steinbacher, M.; Dommen, J.; Prevot, A.S.H.; Fisseha, R.; Weingartner, E.; Frankevich, V.; Zenobi, R.; et al. Identification of polymers as major components of atmospheric organic aerosols. Science 2004, 303, 1659–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehn, M.; Kleist, E.; Junninen, H.; Petaja, T.; Lonn, G.; Schobesberger, S.; Dal Maso, M.; Trimborn, A.; Kulmala, M.; Worsnop, D.R.; et al. Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air. Atmos. Chem. Phys. 2012, 12, 5113–5127. [Google Scholar] [CrossRef] [Green Version]
- Laskin, J.; Laskin, A.; Nizkorodov, S.A.; Roach, P.; Eckert, P.; Gilles, M.K.; Wang, B.B.; Lee, H.J.; Hu, Q.C. Molecular Selectivity of Brown Carbon Chromophores. Environ. Sci. Technol. 2014, 48, 12047–12055. [Google Scholar] [CrossRef] [PubMed]
- Laskin, A.; Laskin, J.; Nizkorodov, S.A. Chemistry of Atmospheric Brown Carbon. Chem. Rev. 2015, 115, 4335–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, R.; Hennigan, C.J.; McMeeking, G.R.; Chuang, W.K.; Robinson, E.S.; Coe, H.; Donahue, N.M.; Robinson, A.L. Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions. Atmos. Chem. Phys. 2013, 13, 7683–7693. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Jang, M. Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight. Atmos. Chem. Phys. 2014, 14, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Bond, T.C. Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion. Geophys. Res. Lett. 2001, 28, 4075–4078. [Google Scholar] [CrossRef] [Green Version]
- Kirchstetter, T.W.; Novakov, T.; Hobbs, P.V. Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon. J. Geophys. Res. Atmos. 2004, 109, 12. [Google Scholar] [CrossRef] [Green Version]
- Lack, D.A.; Langridge, J.M.; Bahreini, R.; Cappa, C.D.; Middlebrook, A.M.; Schwarz, J.P. Brown carbon and internal mixing in biomass burning particles. Proc. Natl. Acad. Sci. USA 2012, 109, 14802–14807. [Google Scholar] [CrossRef] [Green Version]
- Andreae, M.O.; Crutzen, P.J. Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry. Science 1997, 276, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Ng, N.L.; Kroll, J.H.; Chan, A.W.H.; Chhabra, P.S.; Flagan, R.C.; Seinfeld, J.H. Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos. Chem. Phys. 2007, 7, 3909–3922. [Google Scholar] [CrossRef] [Green Version]
- Odum, J.R.; Jungkamp, T.P.W.; Griffin, R.J.; Flagan, R.C.; Seinfeld, J.H. The atmospheric aerosol-forming potential of whole gasoline vapor. Science 1997, 276, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Odum, J.R.; Jungkamp, T.P.W.; Griffin, R.J.; Forstner, H.J.L.; Flagan, R.C.; Seinfeld, J.H. Aromatics, reformulated gasoline, and atmospheric organic aerosol formation. Environ. Sci. Technol. 1997, 31, 1890–1897. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Wang, G.H.; Guo, S.; Zarnora, M.L.; Ying, Q.; Lin, Y.; Wang, W.G.; Hu, M.; Wang, Y. Formation of Urban Fine Particulate Matter. Chem. Rev. 2015, 115, 3803–3855. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, L.; Donahue, N.M.; Pandis, S.N. High formation of secondary organic aerosol from the photo-oxidation of toluene. Atmos. Chem. Phys. 2009, 9, 2973–2986. [Google Scholar] [CrossRef] [Green Version]
- Odum, J.R.; Hoffmann, T.; Bowman, F.; Collins, D.; Flagan, R.C.; Seinfeld, J.H. Gas/particle partitioning and secondary organic aerosol yields. Environ. Sci. Technol. 1996, 30, 2580–2585. [Google Scholar] [CrossRef]
- Lin, P.; Liu, J.M.; Shilling, J.E.; Kathmann, S.M.; Laskin, J.; Laskin, A. Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene. Phys. Chem. Chem. Phys. 2015, 17, 23312–23325. [Google Scholar] [CrossRef]
- Liu, J.M.; Lin, P.; Laskin, A.; Laskin, J.; Kathmann, S.M.; Wise, M.; Caylor, R.; Imholt, F.; Selimovic, V.; Shilling, J.E. Optical properties and aging of light-absorbing secondary organic aerosol. Atmos. Chem. Phys. 2016, 16, 12815–12827. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.Y.; Wang, W.G.; Li, J.L.; Peng, C.; Li, K.; Zhou, L.; Shi, B.; Chen, Y.; Liu, M.Y.; Ge, M.F. Effects of SO2 on optical properties of secondary organic aerosol generated from photooxidation of toluene under different relative humidity conditions. Atmos. Chem. Phys. 2020, 20, 4477–4492. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Wang, W.G.; Ge, M.F.; Li, J.J.; Wang, D. Optical properties of secondary organic aerosols generated by photooxidation of aromatic hydrocarbons. Sci. Rep. 2014, 4, 9. [Google Scholar] [CrossRef]
- Kim, H.; Paulson, S.E. Real refractive indices and volatility of secondary organic aerosol generated from photooxidation and ozonolysis of limonene, alpha-pinene and toluene. Atmos. Chem. Phys. 2013, 13, 7711–7723. [Google Scholar] [CrossRef] [Green Version]
- Moise, T.; Flores, J.M.; Rudich, Y. Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes. Chem. Rev. 2015, 115, 4400–4439. [Google Scholar] [CrossRef]
- Nakayama, T.; Sato, K.; Matsumi, Y.; Imamura, T.; Yamazaki, A.; Uchiyama, A. Wavelength and NOx dependent complex refractive index of SOAs generated from the photooxidation of toluene. Atmos. Chem. Phys. 2013, 13, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.F.; Song, M.J.; Zhao, T.N.; Gunthe, S.S.; Ham, S.H.; He, Y.P.; Qin, Y.M.; Gong, Z.H.; Amorim, J.C.; Bertram, A.K.; et al. Resolving the mechanisms of hygroscopic growth and cloud condensation nuclei activity for organic particulate matter. Nat. Commun. 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.Y.; Huang, D.D.; Li, Z.J.; Liu, Q.Y.; Chan, M.N.; Chan, C.K. Comparison of secondary organic aerosol formation from toluene on initially wet and dry ammonium sulfate particles at moderate relative humidity. Atmos. Chem. Phys. 2018, 18, 5677–5689. [Google Scholar] [CrossRef] [Green Version]
- Tsiligiannis, E.; Hammes, J.; Salvador, C.M.; Mentel, T.F.; Hallquist, M. Effect of NOx on 1,3,5-trimethylbenzene (TMB) oxidation product distribution and particle formation. Atmos. Chem. Phys. 2019, 19, 15073–15086. [Google Scholar] [CrossRef] [Green Version]
- Backstrom, D.; Gunnarsson, A.; Gall, D.; Pei, X.Y.; Johansson, R.; Andersson, K.; Pathak, R.K.; Pettersson, J.B.C. Measurement of the size distribution, volume fraction and optical properties of soot in an 80 kW propane flame. Combust. Flame 2017, 186, 325–334. [Google Scholar] [CrossRef]
- Lopez-Hilfiker, F.D.; Mohr, C.; Ehn, M.; Rubach, F.; Kleist, E.; Wildt, J.; Mentel, T.F.; Lutz, A.; Hallquist, M.; Worsnop, D.; et al. A novel method for online analysis of gas and particle composition: Description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Atmos. Meas. Tech. 2014, 7, 983–1001. [Google Scholar] [CrossRef] [Green Version]
- Aljawhary, D.; Lee, A.K.Y.; Abbatt, J.P.D. High-resolution chemical ionization mass spectrometry (ToF-CIMS): Application to study SOA composition and processing. Atmos. Meas. Tech. 2013, 6, 3211–3224. [Google Scholar] [CrossRef] [Green Version]
- Ziemann, P.J.; Atkinson, R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 2012, 41, 6582–6605. [Google Scholar] [CrossRef]
- Jimenez, P.J.-L. Laboratory Experiments and Modeling for Interpreting Field Studies of Secondary Organic Aerosol Formation Using an Oxidation Flow Reactor; DOE/SC0006035-1; The U.S. Department OF Energy Office of Science Office of Biological and Environmental Research Atmospheric Systems Research Program under Contract NO. DE-SC0006035: Boulder, CO, USA, 2016; pp. 1–46.
- Mao, J.; Ren, X.; Brune, W.H.; Olson, J.R.; Crawford, J.H.; Fried, A.; Huey, L.G.; Cohen, R.C.; Heikes, B.; Singh, H.B.; et al. Airborne measurement of OH reactivity during INTEX-B. Atmos. Chem. Phys. 2009, 9, 163–173. [Google Scholar] [CrossRef] [Green Version]
- DMT. Three-Wavelength Photoacoustic Soot Spectrometer (PASS-3) Operator Manual; DOC-0163 Revision D; Droplet Measurement Technologies, Inc.: Boulder, CO, USA, 2011. [Google Scholar]
- Atkinson, R.; Aschmann, S.M. Rate Constants for the Gas-Phase Reactions of the Oh Radical with a Series of Aromatic-Hydrocarbons at 296+/−2-K. Int. J. Chem. Kinet. 1989, 21, 355–365. [Google Scholar] [CrossRef]
- Simon, L.; Clegg, P.B.; Anthony, S.W. Extended AIM Aerosol Thermodynamics Model. Available online: http://www.aim.env.uea.ac.uk/aim/aim.php (accessed on 8 March 2022).
- Presto, A.A.; Hartz, K.E.H.; Donahue, N.M. Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration. Environ. Sci. Technol. 2005, 39, 7046–7054. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Ng, N.L. Organic Nitrates and Secondary Organic Aerosol (SOA) Formation from Oxidation of Biogenic Volatile Organic Compounds. In Multiphase Environmental Chemistry in the Atmosphere; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2018; Volume 1299, pp. 105–125. [Google Scholar]
- Qi, X.; Zhu, S.P.; Zhu, C.Z.; Hu, J.; Lou, S.R.; Xu, L.; Dong, J.G.; Cheng, P. Smog chamber study of the effects of NOx and NH3 on the formation of secondary organic aerosols and optical properties from photo-oxidation of toluene. Sci. Total Environ. 2020, 727, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Bao, Z.E.; Wu, X.C.; Li, K.W.; Han, L.X.; Zhao, X.Y.; Zhang, X.; Wang, Z.H.; Azzi, M.; Cen, K.F. The effects of humidity and ammonia on the chemical composition of secondary aerosols from toluene/NOx photo-oxidation. Sci. Total Environ. 2020, 728, 10. [Google Scholar] [CrossRef]
- Bao, Z.E.; Xu, H.F.; Li, K.W.; Chen, L.H.; Zhang, X.; Wu, X.C.; Gao, X.; Azzi, M.; Cen, K.F. Effects of NH3 on secondary aerosol formation from toluene/NOx photo-oxidation in different O3 formation regimes. Atmos. Environ. 2021, 261, 11. [Google Scholar] [CrossRef]
- Jang, M.S.; Czoschke, N.M.; Lee, S.; Kamens, R.M. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 2002, 298, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.G.; Inomata, S.; Sato, K.; Ramasamy, S.; Morino, Y.; Enami, S.; Tanimoto, H. Temperature and acidity dependence of secondary organic aerosol formation from alpha-pinene ozonolysis with a compact chamber system. Atmos. Chem. Phys. 2021, 21, 5983–6003. [Google Scholar] [CrossRef]
- Cao, G.; Jang, M. Secondary organic aerosol formation from toluene photooxidation under various NOx conditions and particle acidity. Atmos. Chem. Phys. Discuss. 2008, 2008, 14467–14495. [Google Scholar] [CrossRef] [Green Version]
- McNeill, V.F.; Woo, J.L.; Kim, D.D.; Schwier, A.N.; Wannell, N.J.; Sumner, A.J.; Barakat, J.M. Aqueous-Phase Secondary Organic Aerosol and Organosulfate Formation in Atmospheric Aerosols: A Modeling Study. Environ. Sci. Technol. 2012, 46, 8075–8081. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp.ID | Reacted Toluene (ΔHC) Concentration (ppb) § | NOx | O3 | RH% | NH3 | H2SO4 | OHexpo (Calculated) × 1011 | Remarks |
|---|---|---|---|---|---|---|---|---|
| (NO:NO2 Ratio = 1:1) | (Initial, ppb) | Ramp | (μg m−3) | (cm−3·s) | ||||
| Range | ||||||||
| (Initial to Final, ppb) | ||||||||
| Tol-1 | 1161 | 31 to 362 | 999 | 80 | No | No | 2.8 | NOx ramped |
| Tol-2 | 1064 | 31 to 362 | 999 | 65 | No | No | 2.3 | NOx ramped |
| Tol-3 | 870 | 31 to 362 | 999 | 35 | No | No | 1.6 | NOx ramped |
| Tol-4 | 774 | 31 to 362 | 999 | 20 | No | No | 1.3 | NOx ramped |
| Tol-5 | 1161 | 61 | 999 | 80 | Yes | No | 2.8 | NH3 Ramp. NOx constant |
| Tol-6 | 1161 | 360 | 999 | 80 | Yes | No | 2.8 | NH3 Ramp. NOx constant |
| Tol-7 | 1161 | 30 to 351 | 999 | 80 | No | Yes (22.3) | 2.8 | NOx ramped |
| Tol-8 | 1161 | 30 to 351 | 999 | 65 | No | Yes (22.3) | 2.8 | NOx ramped |
| Tol-9 | 780 | 30 to 351 | 999 | 20 | No | Yes (22.3) | 1.6 | NOx ramped |
| Exp. ID | SOA Type | SOA Mass μg m−3 | Babs.405 Mm−1 | Babs781 Mm−1 | Bscat405 Mm−1 | Bscat781 Mm−1 | MAC405 m2g−1 | MSC405 m2g−1 | OH (Molecules cm−3·s) × 1011; [SOA Aging in Days] | Average Mobility Diameter (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| Tol-1 | NP NR | 552 520 | 11.8 25.2 | 0.320 0.112 | 1259 1258 | 66.5 67.4 | 0.021 0.048 | 2.29 2.38 | 2.8; [1.61] | 65.0 64.7 |
| Tol-2 | NP NR | 372 358 | 8.15 14.5 | 0.000 0.000 | 805 788 | 39.1 38.3 | 0.022 0.040 | 2.13 2.20 | 2.3; [1.33] | 60.8 62.8 |
| Tol-3 | NP NR | 261 223 | 5.31 10.3 | 0.937 0.195 | 516 451 | 22.3 20.3 | 0.020 0.045 | 1.88 1.95 | 1.6; [0.93] | 58.8 57.7 |
| Tol-4 | NP NR | 162 134 | 3.73 6.10 | 0.000 0.042 | 318 258 | 14.0 11.4 | 0.024 0.041 | 2.05 1.77 | 1.3; [0.77] | 57.0 55.0 |
| Tol-5 | NP | 465 | 13.3 | 0.246 | 2958 | 285.8 | 0.029 | 6.39 | 2.8; [1.61] | 94.6 |
| Tol-6 | NR | 432 | 40.1 | 0.025 | 1500 | 0.000 | 0.093 | 3.48 | 2.8; [1.61] | 64.5 |
| Tol-7 | NP NR | 375 416 | 11.5 25.6 | 0.000 0.626 | 1582 1950 | 113.8 139.6 | 0.030 0.061 | 4.19 4.57 | 2.8; [1.61] | 63.9 64.5 |
| Tol-8 | NP NR | 348 321 | 12.4 21.4 | 0.085 0.987 | 1747 1632 | 127.4 117.9 | 0.035 0.068 | 5.05 5.06 | 2.8; [1.61] | 91.2 89.3 |
| Tol-9 | NP NR | 192 182 | 7.86 10.3 | 0.000 0.000 | 807 738 | 55.2 51.4 | 0.041 0.058 | 4.14 4.05 | 1.6; [0.93] | 87.8 88.1 |
| Exp. ID | RH (%) | Avg. SOA Mass (μg m−3) | Avg. NOx/ΔHC | Avg. ALW * (μg m−3) | Avg. SOA Mass # (dry) (μg m−3) | Avg. Babs405 (Mm−1) | Dry SOA MAC405 (m2 g−1) | ALW+SOA MAC405 (m2 g−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NP | NR | NP | NR | NP | NR | NP | NR | NP | NR | NP | NR | NP | NR | ||
| Tol-1 | 80 | 545 | 508 | 0.094 | 0.288 | 167 | 155 | 378 | 353 | 10.3 | 28.0 | 0.027 | 0.079 | 0.019 | 0.055 |
| Tol-2 | 65 | 372 | 342 | 0.094 | 0.288 | 84 | 77 | 289 | 265 | 8.2 | 19.0 | 0.028 | 0.072 | 0.022 | 0.056 |
| Tol -3 | 35 | 261 | 227 | 0.094 | 0.288 | 19 | 17 | 242 | 210 | 4.7 | 10.0 | 0.019 | 0.048 | 0.018 | 0.044 |
| Tol-4 | 20 | 158 | 139 | 0.094 | 0.288 | 6 | 3 | 152 | 136 | 3.3 | 6.5 | 0.022 | 0.048 | 0.021 | 0.047 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, K.; Mishra, H.R.; Pei, X.; Pathak, R.K. Secondary Organic Aerosol (SOA) from Photo-Oxidation of Toluene: 1 Influence of Reactive Nitrogen, Acidity and Water Vapours on Optical Properties. Atmosphere 2022, 13, 1099. https://doi.org/10.3390/atmos13071099
Mitra K, Mishra HR, Pei X, Pathak RK. Secondary Organic Aerosol (SOA) from Photo-Oxidation of Toluene: 1 Influence of Reactive Nitrogen, Acidity and Water Vapours on Optical Properties. Atmosphere. 2022; 13(7):1099. https://doi.org/10.3390/atmos13071099
Chicago/Turabian StyleMitra, Kalyan, Harsh Raj Mishra, Xiangyu Pei, and Ravi Kant Pathak. 2022. "Secondary Organic Aerosol (SOA) from Photo-Oxidation of Toluene: 1 Influence of Reactive Nitrogen, Acidity and Water Vapours on Optical Properties" Atmosphere 13, no. 7: 1099. https://doi.org/10.3390/atmos13071099