
Properties and Atmospheric Oxidation of Norpinic Acid Aerosol
 , and
, and
Abstract
:1. Introduction
2. Experimental Approach and Methods
2.1. Experimental Set Up
2.2. Data Analysis
3. Results and Discussion
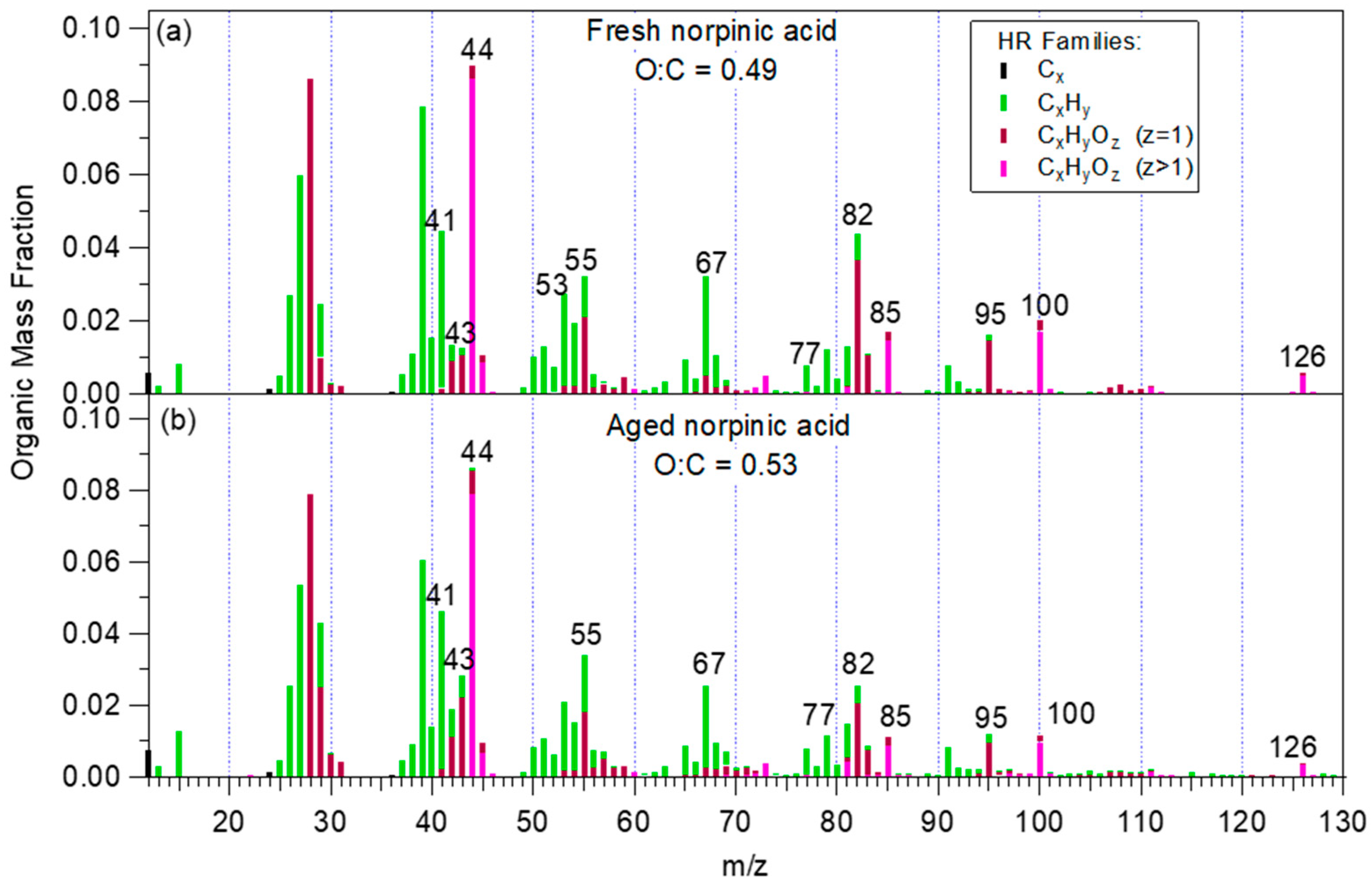
3.1. HR-ToF-AMS Mass Spectrum and Elemental Ratios
3.2. Density and AMS Collection Efficiency
3.3. Norpinic acid Volatility
3.3.1. Using the Thermodenuder Measurements
3.3.2. Isothermal Evaporation of Norpinic Acid in the Chamber
3.4. Chemical Aging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2006, 5, 1053–1123. [Google Scholar] [CrossRef]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar]
- Andersson-Sköld, Y.; Simpson, D. Secondary organic aerosol formation in Northern European model study. J. Geophys. Res. 2001, 106, 7357–7374. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pankow, J.F. Organic atmospheric particulate material. Ann. Rev. Phys. Chem. 2003, 54, 121–140. [Google Scholar] [CrossRef]
- Lee, A.; Goldstein, A.H.; Keywood, M.D.; Gao, S.; Varutbangkul, V.; Bahreini, R.; Ng, N.L.; Flagan, R.C.; Seinfeld, J.H. Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes. J. Geophys. Res. 2006, 111, 1–18. [Google Scholar] [CrossRef]
- Pathak, R.K.; Stanier, C.O.; Donahue, N.M.; Pandis, S.N. Ozonolysis of α-pinene at atmospherically relevant concentrations: Temperature dependence of aerosol mass fractions (yields). J. Geophys. Res. 2007, 112, 1–8. [Google Scholar] [CrossRef]
- Shilling, J.E. Particle mass yield in secondary organic aerosol formed by the dark ozonolysis of α-pinene. Atmos. Chem. Phys. 2008, 8, 2073–2088. [Google Scholar] [CrossRef]
- Kristensen, K.; Jensen, L.N.; Glasius, M.; Bilde, M. The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alphα-pinene. Environ. Sci. Process. Impacts 2017, 19, 1220–1234. [Google Scholar] [CrossRef]
- Friedman, B.; Farmer, D.K. SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes. Atmos. Environ. 2018, 187, 335–345. [Google Scholar] [CrossRef]
- Yu, J.; Flagan, R.C.; Seinfeld, J.H. Identification of products containing -COOH, -OH, and –C=O in atmospheric oxidation of hydrocarbons. Environ. Sci. Technol. 1998, 32, 2357–2370. [Google Scholar] [CrossRef]
- Jang, M.; Kamens, R.M. Newly characterized products and composition of secondary aerosols from the reaction of α-pinene with ozone. Atmos. Environ. 1999, 33, 459–474. [Google Scholar] [CrossRef]
- Glasius, M.; Lahaniati, M.; Calogirou, A.; Di Bella, D.A.; Jensen, N.R.; Hjorth, J.; Kotzias, D.; Larsen, B.R. Carboxylic acids in secondary aerosols from oxidation of cyclic monoterpenes by ozone. Environ. Sci. Technol. 2000, 34, 1001–1010. [Google Scholar] [CrossRef]
- Jaoui, K.; Kamens, R.M. Mass balance of gaseous and particulate products from β-pinene/O3/air in the absence of light and β-pinene/NOx/air in the presence of natural sunlight. J. Atmos. Chem. 2003, 43, 101–141. [Google Scholar] [CrossRef]
- Sato, K.; Jia, T.; Tanabe, K.; Morino, Y.; Kajii, Y.; Imamura, T. Terpenylic acid and nine-carbon multifunctional compounds formed during the aging of β-pinene ozonolysis secondary organic aerosol. Atmos. Environ. 2016, 130, 127–135. [Google Scholar] [CrossRef]
- Bilde, M.; Pandis, S.N. Evaporation rates and vapor pressures of individual aerosol species formed in the atmospheric oxidation of α-pinene. Environ. Sci. Technol. 2001, 35, 3344–3349. [Google Scholar] [CrossRef]
- Steitz, B. Experimental Determination of the Partitioning Coefficient of Nopinone as a Marker Substance in Organic Aerosol. Ph.D. Thesis, Fakultät für Mathematik und Naturwissenschaften, Fach Physik, Bergischen Universität Wuppertal, Wuppertal, Germany, 2012. [Google Scholar]
- Tillmann, R.; Hallquist, M.; Jonsson, Å.M.; Kiendler-Scharr, A.; Saathoff, H.; Iinuma, Y.; Mentel, T.F. Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of α-pinene. Atmos. Chem. Phys. 2010, 10, 7057–7072. [Google Scholar] [CrossRef]
- Müller, M.; Graus, M.; Wisthaler, A.; Hansel, A.; Metzger, A.; Dommen, J.; Baltensperger, U. Analysis of high mass resolution PTR-TOF mass spectra from 1,3,5-trimethylbenzene (TMB) environmental chamber experiments. Atmos. Chem. Phys. 2012, 12, 829–843. [Google Scholar] [CrossRef]
- Yasmeen, F.; Vermeylen, R.; Szmigielski, R.; Iinuma, Y.; Böge, O.; Herrmann, H.; Maenhaut, W.; Claeys, M. Terpenylic acid and related compounds: Precursors for dimers in secondary organic aerosol from the ozonolysis of α- and β-pinene. Atmos. Chem. Phys. 2010, 10, 9383–9392. [Google Scholar] [CrossRef]
- Chacon-Madrid, H.J.; Henry, K.M.; Donahue, N.M. Photo-oxidation of pinonaldehyde at low NOx: From chemistry to organic aerosol formation. Atmos. Chem. Phys. 2013, 13, 3227–3236. [Google Scholar] [CrossRef]
- Yu, J.; Cocker, D.R.; Griffin, R.J.; Flagan, R.C.; Seinfeld, J.H. Gas-phase ozone oxidation of monoterpenes: Gaseous and particulate products. J. Atmos. Chem. 1999, 34, 207–258. [Google Scholar] [CrossRef]
- Kückelmann, U.; Warscheid, B.; Hoffmann, T. On-line characterization of organic aerosols, formed from biogenic precursors using atmospheric pressure chemical ionization mass spectrometry. Anal. Chem. 2000, 72, 1905–1912. [Google Scholar] [CrossRef]
- Iinuma, Y.; Boge, O.; Gnauk, T.; Herrmann, H. Aerosol-chamber study of the α-pinene/O3 reaction: Influence of particle acidity on aerosol yields and products. Atmos. Environ. 2004, 38, 761–773. [Google Scholar] [CrossRef]
- Ma, Y.; Russell, A.T.; Marston, G. Mechanisms for the formation of secondary organic aerosol components from the gas-phase ozonolysis of α-pinene. Phys. Chem. Chem. Phys. 2008, 10, 4294–4312. [Google Scholar] [CrossRef] [PubMed]
- Camredon, M.; Hamilton, J.F.; Alam, M.S.; Wyche, K.P.; Carr, T.; White, I.R.; Monks, P.S.; Rickard, A.R.; Bloss, W.J. Distribution of gaseous and particulate organic composition during dark α-pinene ozonolysis. Atmos. Chem. Phys. 2010, 10, 2893–2917. [Google Scholar] [CrossRef]
- Geddes, S.; Nichols, B.; Todd, K.; Zahardis, J.; Petrucci, G.A. Near-infrared laser desorption/ionization aerosol mass spectrometry for measuring organic aerosol at atmospherically relevant aerosol mass loadings. Atmos. Meas. Tech. 2010, 3, 1175–1183. [Google Scholar] [CrossRef]
- Boy, M.; Petäjä, T.; Dal Maso, M.; Rannik, Ü.; Rinne, J.; Aalto, P.; Laaksonen, A.; Vaattovaara, P.; Joutsensaari, J.; Hoffmann, T.; et al. Overview of the field measurement campaign in Hyytiälä, August 2001 in the framework of the EU project OSOA. Atmos. Chem. Phys. 2004, 4, 657–678. [Google Scholar] [CrossRef]
- Plewka, A.; Gnauk, T.; Bruggemann, E.; Herrmann, H. Biogenic contributions to the chemical composition of airborne particles in a coniferous forest in Germany. Atmos. Environ. 2006, 40, S103–S115. [Google Scholar] [CrossRef]
- Kourtchev, I.; Ruuskanen, T.M.; Keronen, P.; Sogacheva, L.; Dal Maso, M.; Reissell, A.; Chi, X.; Vermeylen, R.; Kulmala, M.; Maenhaut, W.; et al. Determination of isoprene and α-/β-pinene oxidation products in boreal forest aerosols from Hyytiälä, Finland: Diel variations and possible link with particle formation events. Plant Biol. 2008, 10, 138–149. [Google Scholar] [CrossRef]
- Fu, P.; Kawamura, K.; Chen, J.; Barrie, L.A. Isoprene, monoterpene, and sesquiterpene oxidation products in the High Arctic aerosols during late winter to early summer. Environ. Sci. Technol. 2009, 43, 4022–4028. [Google Scholar] [CrossRef]
- Fu, K.; Kawamura, K.; Pavuluri, C.M.; Jing, C.; Swaminathan, T. Contributions of isoprene, α/β-pinene and β-caryophyllene to secondary organic aerosol in tropical India. Low Temp. Sci. 2010, 68, 79–88. [Google Scholar]
- Fu, K.; Kawamura, K. Diurnal variations of polar organic tracers in summer forest aerosols: A case study of a Quercus and Picea mixed forest in Hokkaido, Japan. Geochem. J. 2011, 45, 297–308. [Google Scholar] [CrossRef]
- Yasmeen, F.; Szmigielski, R.; Vermeylen, R.; Gomez-Gonzalez, Y.; Surratt, J.D.; Chan, A.W.H.; Seinfeld, J.H.; Maenhaut, W.; Claeys, M. Mass spectrometric characterization of isomeric terpenoic acids from the oxidation of α-pinene, β-pinene, d-limonene and Δ3-carene in fine forest aerosol. J. Mass. Spectrom. 2011, 46, 425–442. [Google Scholar] [CrossRef]
- Li, L.; Dai, D.; Deng, S.; Feng, J.; Zhao, M.; Wu, J.; Liu, L.; Yang, X.; Wu, S.; Qi, H.; et al. Concentration, distribution and variation of polar organic aerosol tracers in Ya’an, a middle-sized city in western China. Atmos. Res. 2013, 120, 29–42. [Google Scholar] [CrossRef]
- Kołodziejczyk, A.; Pyrcz, P.; Pobudkowska, A.; Błaziak, K.; Szmigielski, R. Physicochemical properties of pinic, pinonic, norpinic, and norpinonic acids as relevant α-pinene oxidation products. J. Phys. Chem. B 2019, 123, 8261–8267. [Google Scholar] [CrossRef]
- Yasmeen, F.; Vermeylen, R.; Maurin, N.; Perraudin, E.; Doussin, J.F.; Claeys, M. Characterisation of tracers for aging of α-pinene secondary organic aerosol using liquid chromatography/negative ion electrospray ionisation mass spectrometry. Environ. Chem. 2012, 9, 236–246. [Google Scholar] [CrossRef]
- Kenseth, C.M.; Huang, Y.; Zhao, R.; Dalleska, N.F.; Hethcoxa, J.C.; Stoltz, B.M.; Seinfeld, J.H. Synergistic O3 + OH oxidation pathway to extremely low-volatility dimers revealed in β-pinene secondary organic aerosol. PNAS 2018, 115, 8301–8306. [Google Scholar] [CrossRef] [PubMed]
- Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ. 1997, 31, 81–104. [Google Scholar] [CrossRef]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.G.; Pilling, M.J. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef]
- Kostenidou, E.; Karnezi, E.; Kołodziejczyk, A.; Szmigielski, R.; Pandis, S.N. Physical and chemical properties of 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) aerosol. Environ. Sci. Technol. 2018, 52, 1150–1155. [Google Scholar] [CrossRef]
- Barmet, P.; Dommen, J.; DeCarlo, P.F.; Tritscher, T.; Praplan, A.P.; Platt, S.M.; Prevot, A.S.H.; Donahue, N.M.; Baltensperger, U. OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber. Atmos. Meas. Tech. 2012, 5, 647–656. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- An, W.J.; Pathak, R.K.; Lee, B.H.; Pandis, S.N. Aerosol volatility measurement using an improved thermodenuder: Application to secondary organic aerosol. J. Aerosol Sci. 2007, 38, 305–314. [Google Scholar] [CrossRef]
- Louvaris, E.E.; Karnezi, E.; Kostenidou, E.; Kaltsonoudis, C.; Pandis, S.N. Estimation of the volatility distribution of organic aerosol combining thermodenuder and isothermal dilution measurements. Atmos. Meas. Tech. 2017, 10, 3909–3918. [Google Scholar] [CrossRef]
- Riipinen, I.; Pierce, J.R.; Donahue, N.M.; Pandis, S.N. Equilibration time scales of organic aerosol inside thermodenuders: Evaporation kinetics versus thermodynamics. Atmos. Environ. 2010, 44, 597–607. [Google Scholar] [CrossRef]
- Wang, N.; Jorga, S.D.; Pierce, J.R.; Donahue, N.M.; Pandis, S.N. Particle wall-loss correction methods in smog chamber experiments. Atmos. Meas. Tech. 2018, 11, 6577–6588. [Google Scholar] [CrossRef]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef]
- Aiken, A.C.; Decarlo, P.F.; Kroll, J.H.; Worsnop, D.R.; Huffman, J.A.; Docherty, K.S.; Ulbrich, I.M.; Mohr, C.; Kimmel, J.R.; Sueper, D.; et al. O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with High Resolution Time-of-Flight Aerosol Mass Spectrometry. Environ. Sci. Technol. 2008, 42, 4478–4485. [Google Scholar] [CrossRef]
- Kostenidou, E.; Pathak, R.K.; Pandis, S.N. An algorithm for the calculation of secondary organic aerosol density combining AMS and SMPS data. Aerosol Sci. Technol. 2007, 41, 1002–1010. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive matrix factorization – a nonnegative factor model with optimal utilization of error-estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prévôt, A.S.H. Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. 2007, 7, 1503–1522. [Google Scholar]
- Ulbrich, I.M.; Canagaratna, M.R.; Zhang, Q.; Worsnop, D.R.; Jimenez, J.L. Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. 2009, 9, 2891–2918. [Google Scholar] [CrossRef]
- Kostenidou, E.; Lee, B.H.; Engelhart, G.J.; Pierce, J.R.; Pandis, S.N. Mass spectra deconvolution of low, medium and high volatility biogenic secondary organic aerosol. Environ. Sci. Technol. 2009, 43, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.H.; Hamilton, J.F.; Allan, J.D.; Langford, B.; Oram, D.E.; Chen, Q.; Docherty, K.; Farmer, D.K.; Jimenez, J.L.; Ward, M.W.; et al. Evidence for a significant proportion of secondary organic aerosol from isoprene above a maritime tropical forest. Atmos. Chem. Phys. 2011, 11, 1039–1050. [Google Scholar]
- Slowik, J.G.; Brook, J.; Chang, R.Y.W.; Evans, G.J.; Hayden, K.; Jeong, C.H.; Li, S.M.; Liggio, J.; Liu, P.S.K.; McGuire, M.; et al. Photochemical processing of organic aerosol at nearby continental sites: Contrast between urban plumes and regional aerosol. Atmos. Chem. Phys. 2011, 11, 2991–3006. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Farmer, D.K.; Rizzo, L.V.; Pauliquevis, T.; Kuwata, M.; Karl, T.G.; Guenther, A.; Allan, J.D.; Coe, H.; Andreae, M.O.; et al. Submicron particle mass concentrations and sources in the Amazonian wet season (AMAZE-08). Atmos. Chem. Phys. 2015, 15, 3687–3701. [Google Scholar]
- Kostenidou, E.; Florou, K.; Kaltsonoudis, C.; Tsiflikiotou, M.; Vratolis, S.; Eleftheriadis, K.; Pandis, S.N. Sources and chemical characterization of organic aerosol during the summer in the eastern Mediterranean. Atmos. Chem. Phys. 2015, 15, 11355–11371. [Google Scholar] [CrossRef]
- Budisulistiorini, S.H.; Canagaratna, M.R.; Croteau, P.L.; Marth, W.J.; Baumann, K.; Edgerton, E.S.; Shaw, S.L.; Knipping, E.M.; Worsnop, D.R.; Jayne, J.T.; et al. Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in downtown Atlanta, Georgia, using the Aerodyne Aerosol Chemical Speciation Monitor. Environ. Sci. Technol. 2013, 47, 5686–5694. [Google Scholar] [CrossRef]
- Xu, L.; Guo, H.; Boyd, C.M.; Klein, M.; Bougiatioti, A.; Cerully, K.M.; Hite, J.R.; Isaacman-VanWertz, G.; Kreisberg, N.M.; Knote, C.; et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the Southeastern United States. PNAS 2015, 112, 37–42. [Google Scholar] [CrossRef]
- Milic, A.; Mallet, M.D.; Cravigan, L.T.; Alroe, J.; Ristovski, Z.D.; Selleck, P.; Lawson, S.J.; Ward, J.; Desservettaz, M.J.; Paton-Walsh, C.; et al. Biomass burning and biogenic aerosols in northern Australia during the SAFIRED campaign. Atmos. Chem. Phys. 2017, 17, 3945–3961. [Google Scholar] [CrossRef]
- Lin, Y.H.; Zhang, Z.; Docherty, K.S.; Zhang, H.; Budisulistiorini, S.H.; Rubitschun, C.L.; Shaw, S.L.; Knipping, E.M.; Edgerton, E.S.; Kleindienst, T.E.; et al. Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic compounds. Environ. Sci. Technol. 2011, 46, 250–258. [Google Scholar] [CrossRef]
- Wang, N.; Kostenidou, E.; Donahue, N.M.; Pandis, S.N. Multi-generation chemical aging of α-pinene ozonolysis products by reactions with OH. Atmos. Chem. Phys. 2018, 18, 3589–3601. [Google Scholar] [CrossRef]
- Tasoglou, A.; Pandis, S.N. Formation and chemical aging of secondary organic aerosol during the β-caryophyllene oxidation. Atmos. Chem. Phys. 2015, 15, 6035–6046. [Google Scholar] [CrossRef]
- Kuwata, M.; Zorn, S.R.; Martin, S.T. Using elemental ratios to predict the density of organic material composed of carbon, hydrogen, and oxygen. Environ. Sci. Technol. 2012, 46, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Prinn, R.; Huang, J.; Weiss, R.; Cunnold, D.; Fraser, P.; Simmonds, P.; McCulloch, A.; Harth, C.; Reimann, S.; Salameh, P.; et al. Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. Geophys. Res. Lett. 2005, 32, L07809. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Facility | ICE-FORTH | CMU | ||||
|---|---|---|---|---|---|---|
| Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 | Exp. 6 | |
| Initial norpinic acid (μg m−3) | 73.3 | 84.3 | 11.6 | 33.1 | 47 | 41 |
| Initial (NH4)2SO4 (μg m−3) | 31.6 a | 82 b | NA | NA | 44 b | 34 b |
| AMS Collection Efficiency | 0.38 ± 0.05 | 0.4 ± 0.03 | 0.4 ± 0.03 | 0.24 ± 0.03 | 0.6 ± 0.07 | 0.75 ± 0.12 |
| Density (g cm−3) | 1.29 ± 0.02 | 1.32 ± 0.03 | 1.3 ± 0.02 | 1.29 ± 0.02 | ΝA | ΝA |
| Butanol-d9 (ppb) | 0 | 82 | 182 | 17 | NA | |
| Equivalent OH exposure (days) c | 0 | 5 | 2 | 0 | 0.6 | NA |
| T (°C) | 20.7 ± 0.5 | 29.1 ± 0.7 | 22.8 ± 3.7 | 23.6 ± 1.3 | 27.3 ± 1.8 | 28.1 ± 1.4 |
| RH (%) | 11 ± 0.3 | 9.6 ± 0.7 | 12.8 ± 3.5 | 16.5 ± 5.3 | 5 ± 0.8 | 6 ± 0.9 |
| Angle Theta in Degrees (R2) | Reference | |
|---|---|---|
| Factor 82 | 48.6 (0.35) | Robinson et al. [54] |
| IEPOX OA | 73.5 (0.01) | Budisulistiorini et al. [58] |
| Isoprene-OA | 31.9 (0.69) | Xu et al. [59] |
| Biogenic OA | 26.3 (0.78) | Kostenidou et al. [57] |
| MBTCA | 24.3 (0.82) | Kostenidou et al. [40] |
| α-pinene + O3 SOA | 27.7 (0.76) | Wang et al. [62] |
| β-carryophylene + O3 SOA | 33.8 (0.65) | Tasoglou et al. [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostenidou, E.; Jorga, S.; Kodros, J.K.; Florou, K.; Kołodziejczyk, A.; Szmigielski, R.; Pandis, S.N. Properties and Atmospheric Oxidation of Norpinic Acid Aerosol. Atmosphere 2022, 13, 1481. https://doi.org/10.3390/atmos13091481
Kostenidou E, Jorga S, Kodros JK, Florou K, Kołodziejczyk A, Szmigielski R, Pandis SN. Properties and Atmospheric Oxidation of Norpinic Acid Aerosol. Atmosphere. 2022; 13(9):1481. https://doi.org/10.3390/atmos13091481
Chicago/Turabian StyleKostenidou, Evangelia, Spiro Jorga, John K. Kodros, Kalliopi Florou, Agata Kołodziejczyk, Rafal Szmigielski, and Spyros N. Pandis. 2022. "Properties and Atmospheric Oxidation of Norpinic Acid Aerosol" Atmosphere 13, no. 9: 1481. https://doi.org/10.3390/atmos13091481
APA StyleKostenidou, E., Jorga, S., Kodros, J. K., Florou, K., Kołodziejczyk, A., Szmigielski, R., & Pandis, S. N. (2022). Properties and Atmospheric Oxidation of Norpinic Acid Aerosol. Atmosphere, 13(9), 1481. https://doi.org/10.3390/atmos13091481