
Theoretical Study on the Mechanisms, Kinetics, and Toxicity Evaluation of OH-Initiated Atmospheric Oxidation Reactions of Coniferyl Alcohol



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methods
2.1. Electronic Structures Calculations
2.2. Rate Constant Calculations
2.3. Toxicity Assessment
3. Results and Discussion
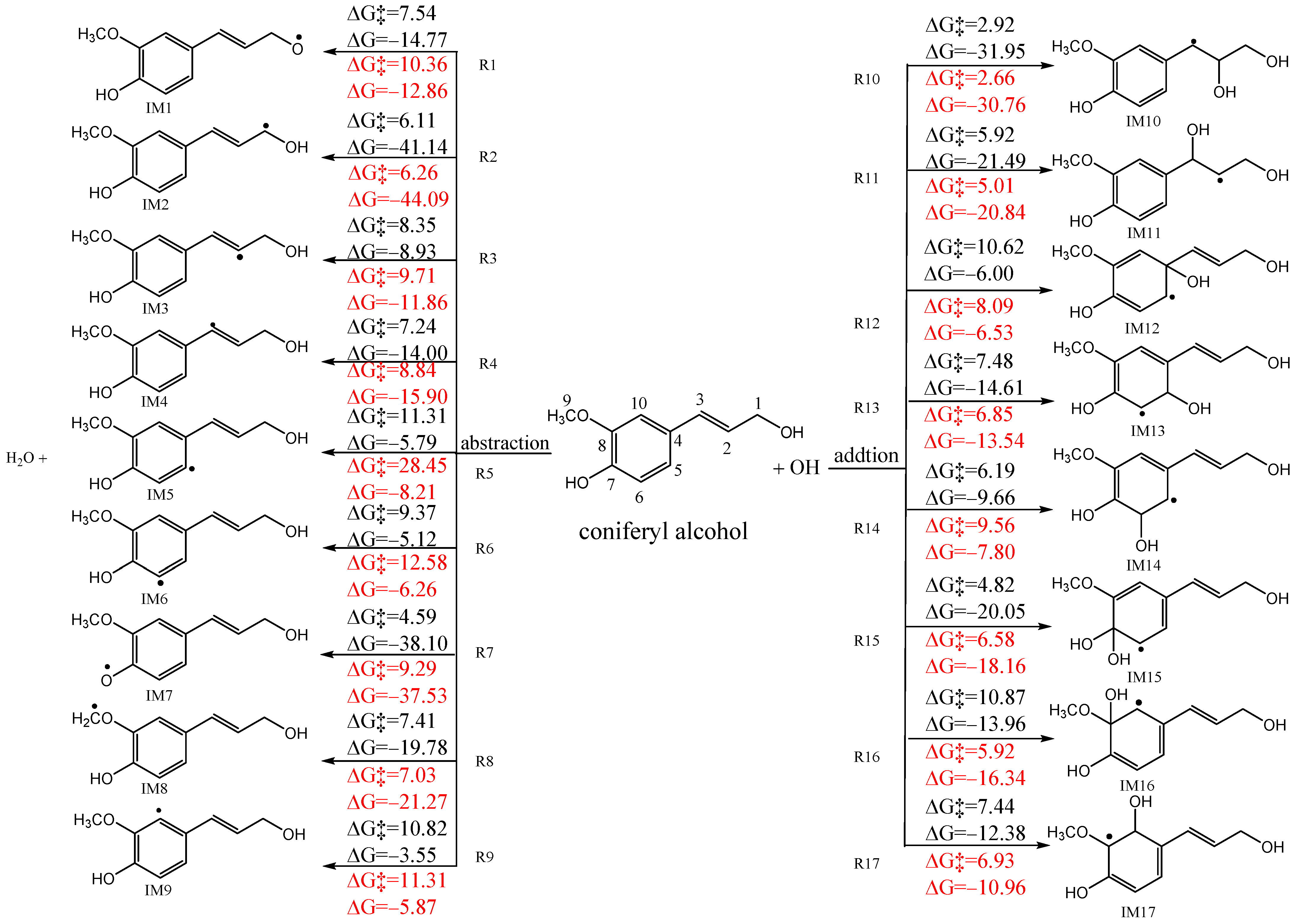
3.1. Preliminary Reactions of OH Radicals with CA
3.1.1. H-Abstraction Mechanisms
3.1.2. OH-Addition Mechanisms
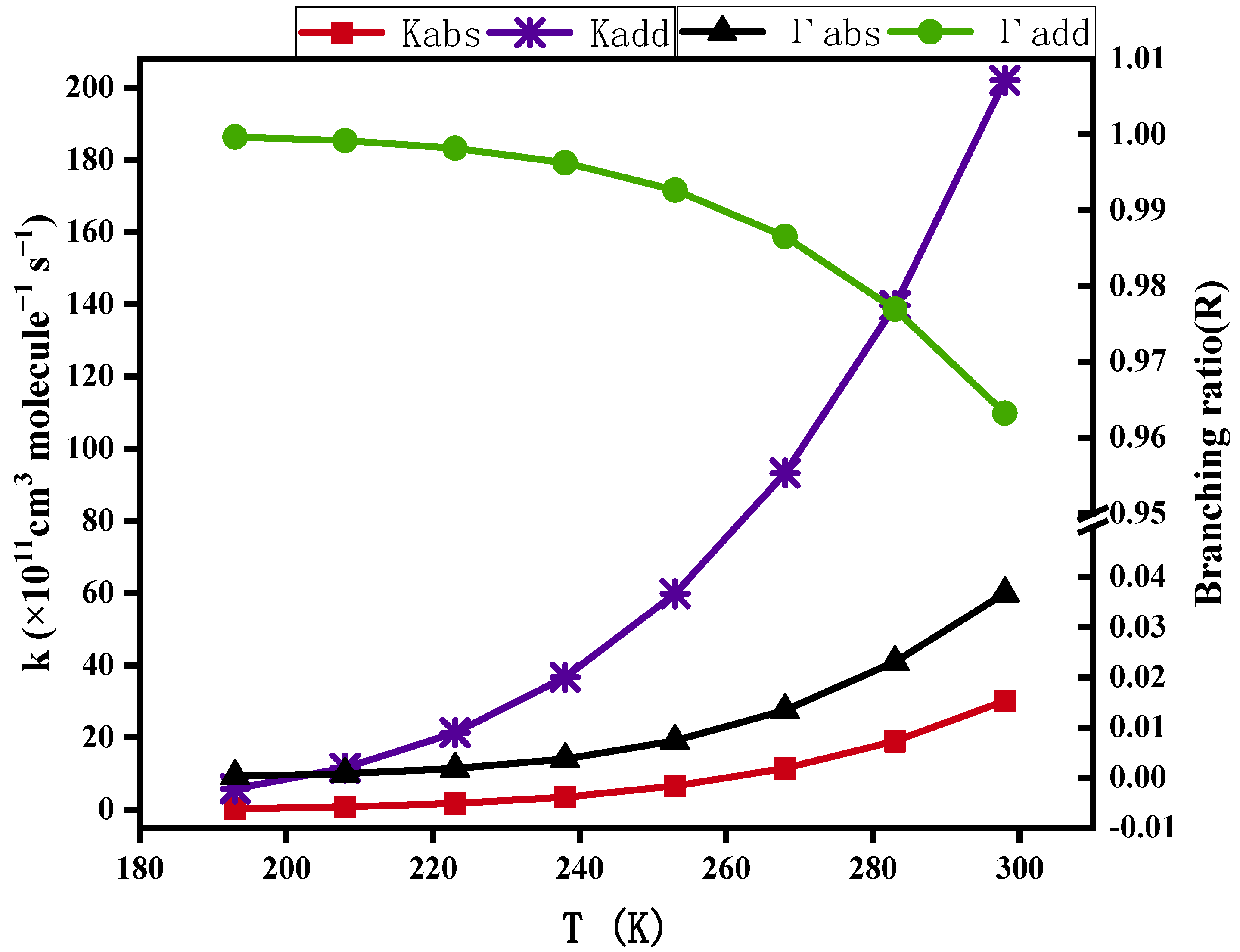
3.1.3. Pre-Reactive Complexes and Kinetics
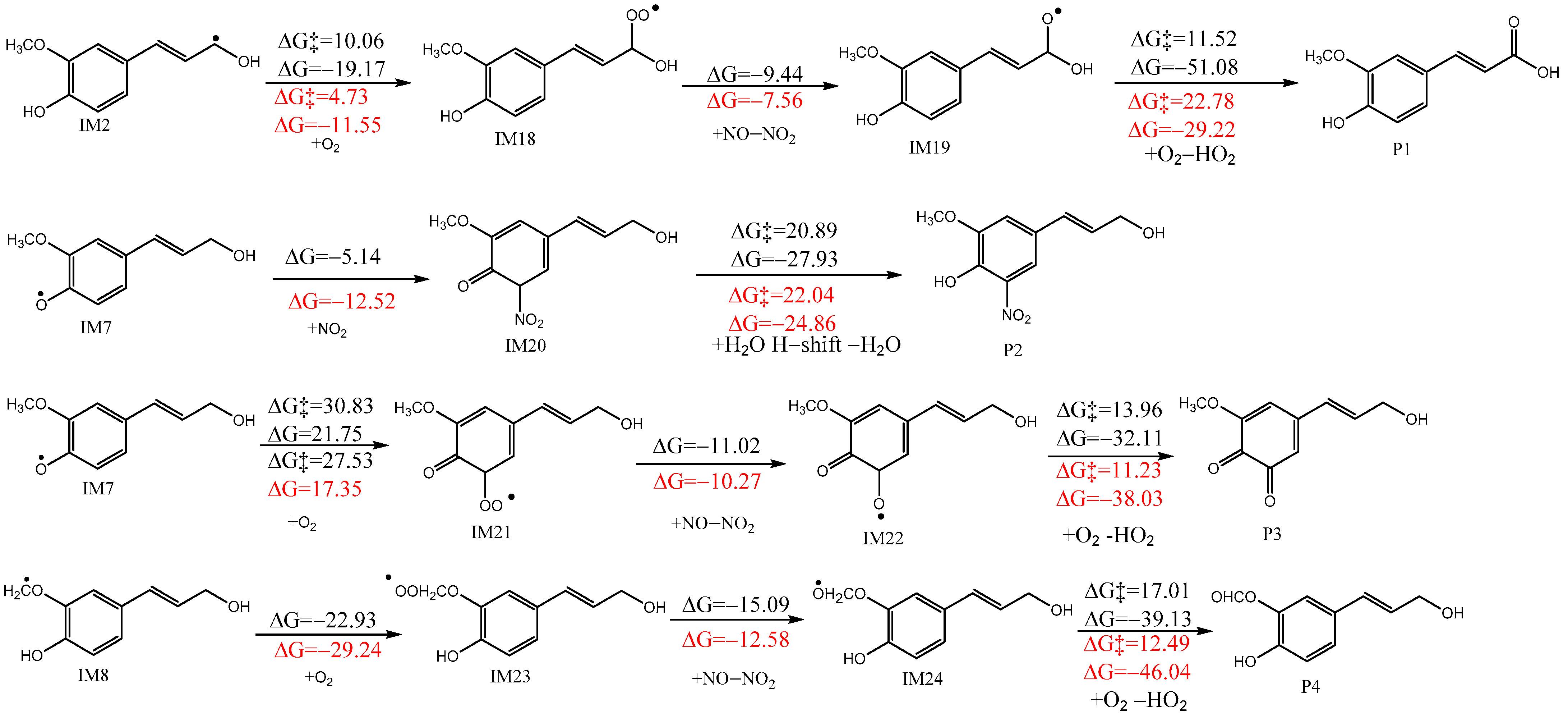
3.2. Subsequent Reactions of IM2, IM7, and IM8
3.2.1. Reaction with NO2
3.2.2. Reactions with O2
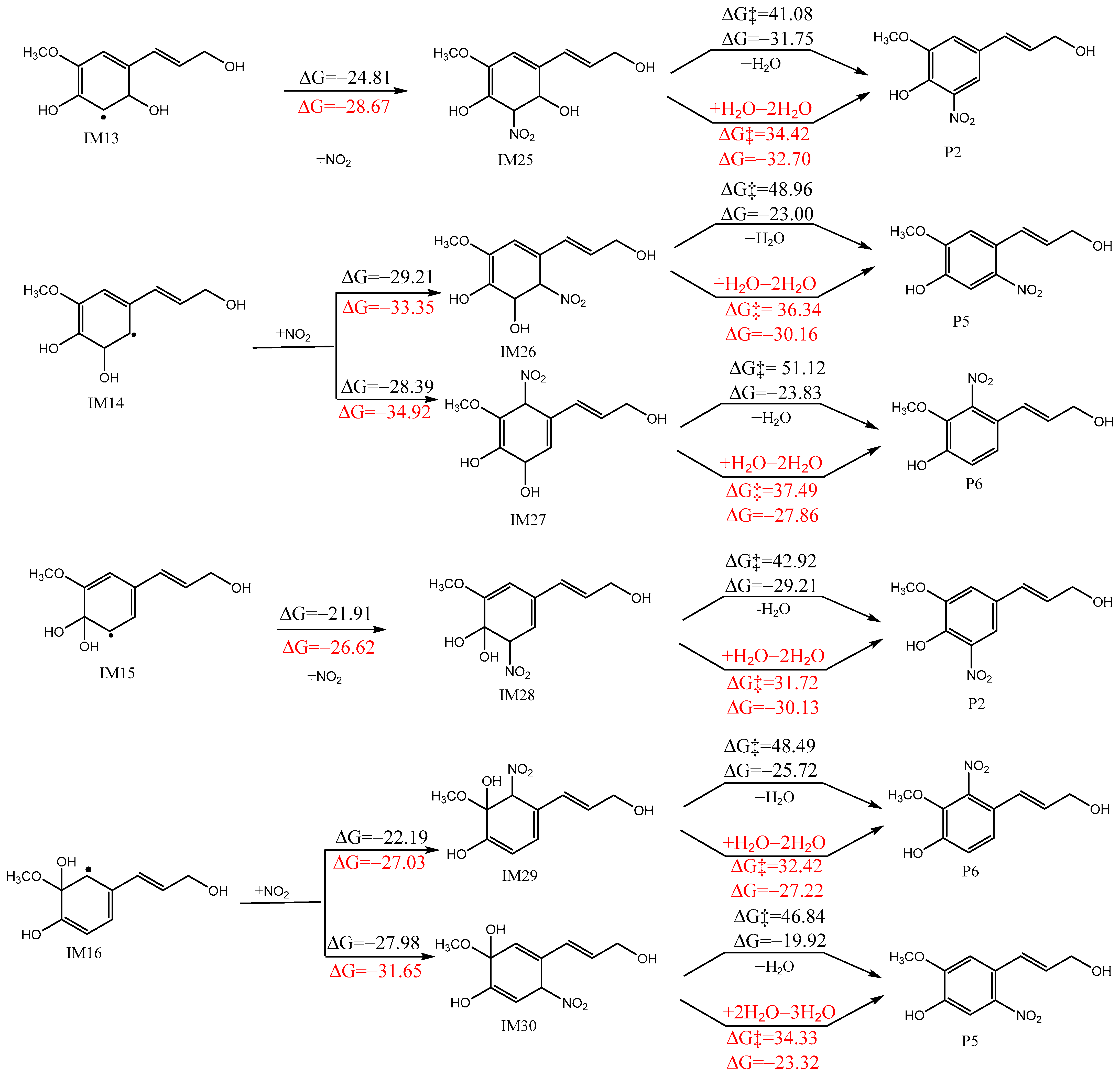
3.3. Subsequent Reactions of IM10–IM16
3.3.1. Reactions with NO2
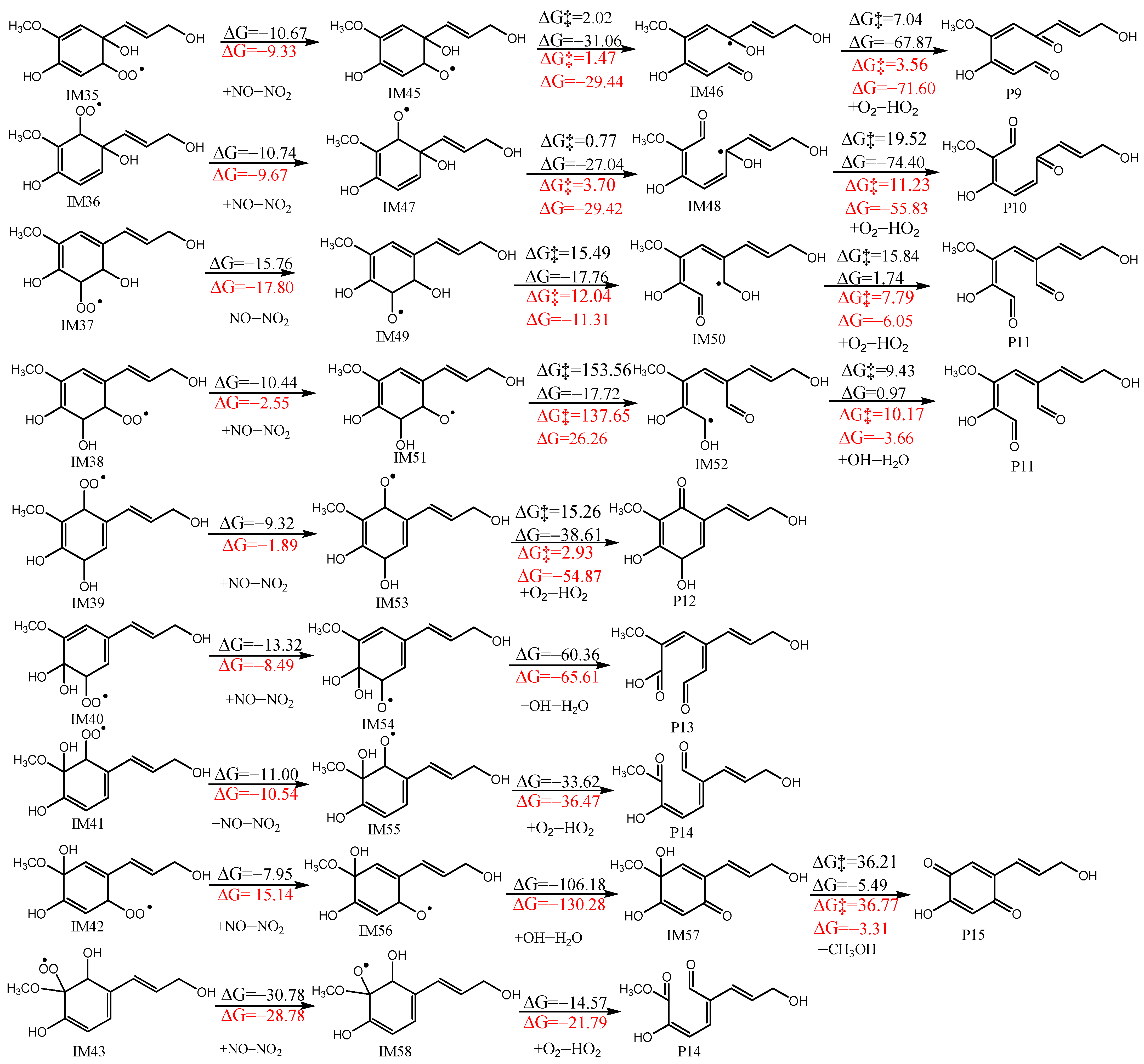
3.3.2. Reactions with O2
3.4. Toxicity Assessment
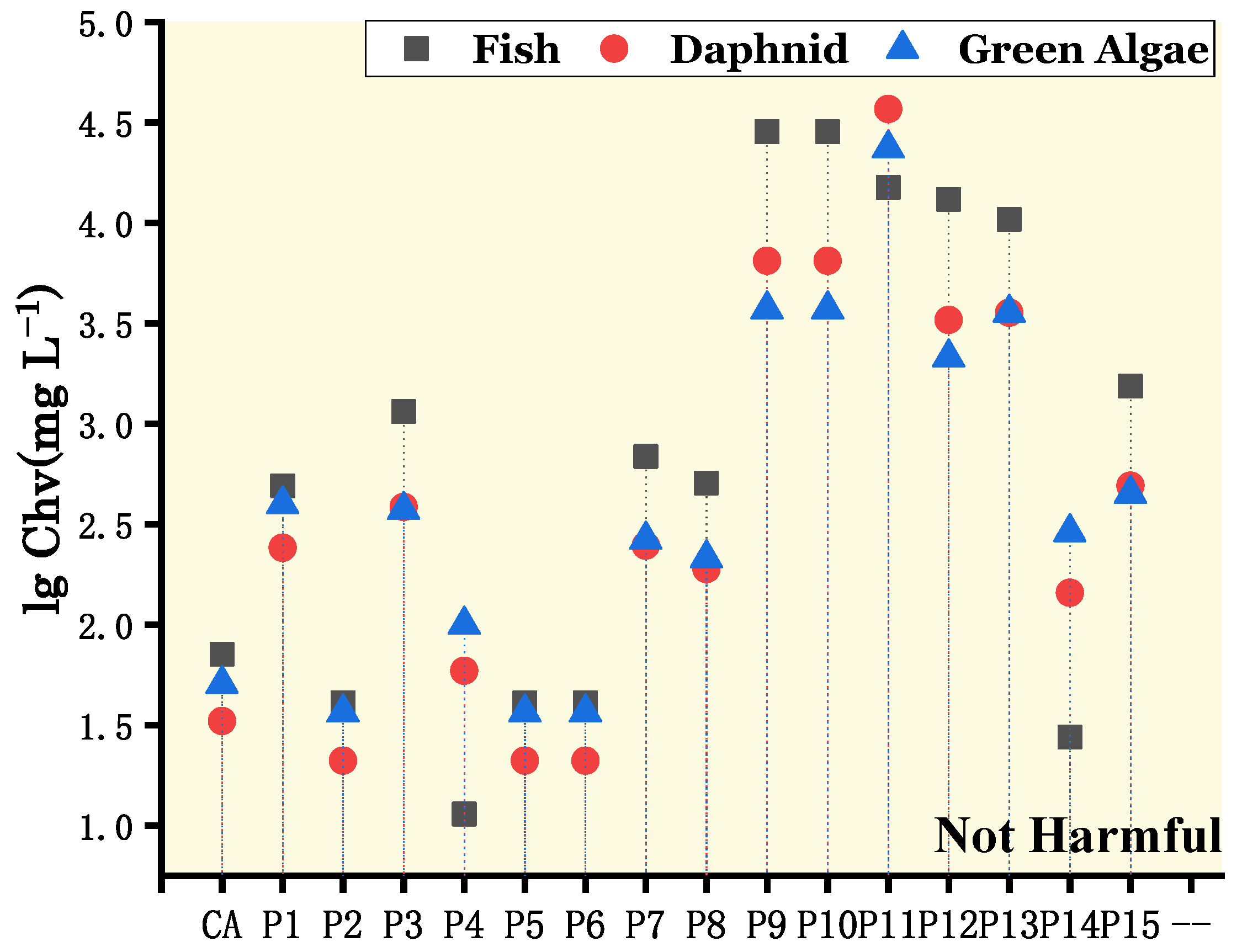
3.4.1. Acute and Chronic Toxicity of CA to Aquatic Organisms
3.4.2. The Toxicity of the Products in the NO2 and O2 Addition Pathways
3.4.3. Developmental Toxicity and Mutagenicity
4. Conclusions
- (1)
- All of the preliminary OH-addition reactions and H-abstraction reactions of CA have been studied, and the role of the atmospheric liquid phase was examined. Except for the R8 pathway, the energy barrier of initial H-abstraction reactions decreased due to the atmospheric liquid phase, whereas that for most of the initial OH-addition reactions decreased, except for R14 and R15. Furthermore, the reaction order of different pathways changed, with the long-branched chain being more susceptible to initial reactions than the benzene ring. The subsequent reactions of CA with O2/NOx were studied using favorable pathways IM2, IM7, IM8, and IM10–IM16.
- (2)
- In the atmospheric liquid phase, the total reaction rate constants for the preliminary reactions of CA with OH radicals at 298 K were 2.32 × 10−9 cm3 molecule−1 s−1 and 9.44 × 109 s−1M−1, with half-lives of 8.3 × 10−2 h, 5.83 × 103 h, and 9.27 × 102 h, respectively. The initial reaction rate constant was positively correlated with temperature. OH-addition reactions played a significant role in the initial reaction with OH radicals in the atmospheric liquid phase, with branching ratios of H-abstraction reactions and OH-addition reactions being 3.68% and 97.69%, respectively.
- (3)
- Ecotoxicity evaluation revealed that CA and its products had similar levels of toxicity, which were non-toxic. However, all these products had developmental toxicity, and most of them were also mutagenic. Therefore, the ecotoxicity testing of the CA and degradation products and the effects on human health should be closely monitored.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baz, K.; Cheng, J.; Xu, D.; Abbas, K.; Ali, I.; Ali, H.; Fang, C. Asymmetric impact of fossil fuel and renewable energy consumption on economic growth: A nonlinear technique. Energy 2021, 226, 120357. [Google Scholar] [CrossRef]
- Zheng, S.; Zhou, X.; Tan, Z.; Liu, C.; Hu, H.; Yuan, H.; Peng, S.; Cai, X. Assessment of the global energy transition: Based on trade embodied energy analysis. Energy 2023, 273, 127274. [Google Scholar] [CrossRef]
- Glushkov, D.O.; Nyashina, G.S.; Anand, R.; Strizhak, P.A. Composition of gas produced from the direct combustion and pyrolysis of biomass. Process Saf. Environ. Prot. 2021, 156, 43–56. [Google Scholar] [CrossRef]
- Bildirici, M.; Özaksoy, F. An analysis of biomass consumption and economic growth in transition countries. Econ. Res.-Ekon. Istraživanja 2018, 31, 386–405. [Google Scholar] [CrossRef] [Green Version]
- Gnanasekaran, L.; Priya, A.K.; Thanigaivel, S.; Hoang, T.K.; Soto-Moscoso, M. The conversion of biomass to fuels via cutting-edge technologies: Explorations from natural utilization systems. Fuel 2023, 331, 125668. [Google Scholar] [CrossRef]
- Levine, J. Biomass burning: The cycling of gases and particulates from the biosphere to the atmosphere. Treatise Geochem. 2003, 4, 347. [Google Scholar]
- Lelieveld, J.; Crutzen, P.J.; Ramanathan, V.; Andreae, M.O.; Brenninkmeijer, C.A.M.; Campos, T.; Cass, G.R.; Dickerson, R.R.; Fischer, H.; De Gouw, J.A.; et al. The Indian Ocean experiment: Widespread air pollution from South and Southeast Asia. Science 2001, 291, 1031–1036. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Li, G.; Pongpiachan, S.; Wang, Q.; Han, Y.; Cao, J.; Tipmanee, D.; Palakun, J.; Aukkaravittayapun, S.; Surapipith, V.; et al. Quantifying the contributions of local emissions and regional transport to elemental carbon in Thailand. Environ. Pollut. 2020, 262, 114272. [Google Scholar] [CrossRef]
- Kocbach Bølling, A.; Pagels, J.; Yttri, K.E.; Barregard, L.; Sallsten, G.; Schwarze, P.E.; Boman, C. Health effects of residential wood smoke particles: The importance of combustion conditions and physicochemical particle properties. Part. Fibre Toxicol. 2009, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, R.C. The chemical composition of wood. Chem. Solid Wood 1984, 207, 57–126. [Google Scholar]
- Yang, B.; Zhang, H.; Wang, Y.; Zhang, P.; Shu, J.; Sun, W.; Ma, P. Experimental and theoretical studies on gas-phase reactions of NO3 radicals with three methoxyphenols: Guaiacol, creosol, and syringol. Atmos. Environ. 2016, 125, 243–251. [Google Scholar] [CrossRef]
- Michałowicz, J.; Stufka-Olczyk, J.; Milczarek, A.; Michniewicz, M. Analysis of annual fluctuations in the content of phenol, chlorophenols and their derivatives in chlorinated drinking waters. Environ. Sci. Pollut. Res. 2011, 18, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, S.B.; Miller, D.J.; Barkley, R.M.; Krieger, M.S. Identification of methoxylated phenols as candidate tracers for atmospheric wood smoke pollution. Environ. Sci. Technol. 1988, 22, 1191–1196. [Google Scholar] [CrossRef]
- An, Z.; Sun, J.; Han, D.; Mei, Q.; Wei, B.; Wang, X.; He, M. Theoretical study on the mechanisms, kinetics and ecotoxicity assessment of OH-initiated reactions of guaiacol in atmosphere and wastewater. Sci. Total Environ. 2019, 685, 729–740. [Google Scholar] [CrossRef]
- Lauraguais, A.; Bejan, I.; Barnes, I.; Wiesen, P.; Coeur, C. Rate coefficients for the gas-phase reactions of hydroxyl radicals with a series of methoxylated aromatic compounds. J. Phys. Chem. A 2015, 119, 6179–6187. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Chen, T.; Liu, J.; He, H. Rate constant and secondary organic aerosol formation from the gas-phase reaction of eugenol with hydroxyl radicals. Atmos. Chem. Phys. 2019, 19, 2001–2013. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Sun, J.; Mei, Q.; An, Z.; Wang, X.; He, M. Theoretical study on gas-phase reactions of nitrate radicals with methoxyphenols: Mechanism, kinetic and toxicity assessment. Environ. Pollut. 2018, 243, 1772–1780. [Google Scholar] [CrossRef]
- Zein, A.E.; Coeur, C.; Obeid, E.; Lauraguais, A.; Fagniez, T. Reaction kinetics of catechol (1,2-benzenediol) and guaiacol (2-methoxyphenol) with ozone. J. Phys. Chem. A 2015, 119, 6759–6765. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Wang, Y.; Shu, J.; Zhang, P.; Ma, P.; Li, Z. Gas-phase reactions of methoxyphenols with NO3 radicals: Kinetics, products, and mechanisms. J. Phys. Chem. A 2016, 120, 1213–1221. [Google Scholar] [CrossRef]
- Lv, Y.; Cheng, X.; Wu, D.; Du, G.; Zhou, J.; Chen, J. Improving bioconversion of eugenol to coniferyl alcohol by in situ eliminating harmful H2O2. Bioresour. Technol. 2018, 267, 578–583. [Google Scholar] [CrossRef]
- Nolte, C.G.; Schauer, J.J.; Cass, G.R.; Simoneit, B.R. Highly polar organic compounds present in wood smoke and in the ambient atmosphere. Environ. Sci. Technol. 2001, 35, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, Y. Kinetic study on the heterogeneous degradation of coniferyl alcohol by OH radicals. Chemosphere 2020, 241, 125088. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wen, X.; Wu, B. Heterogeneous reaction of coniferyl alcohol adsorbed on silica particles with NO3 radicals. Atmos. Pollut. Res. 2017, 8, 514–520. [Google Scholar] [CrossRef]
- Net, S.; Alvarez, E.G.; Gligorovski, S.; Wortham, H. Heterogeneous reactions of ozone with methoxyphenols, in presence and absence of light. Atmos. Environ. 2011, 45, 3007–3014. [Google Scholar] [CrossRef]
- Comes, F.J. Recycling in the Earth’s Atmosphere: The OH Radical—Its Importance for the Chemistry of the Atmosphere and the Determination of Its Concentration. Angew. Chem. Int. Ed. Engl. 1994, 33, 1816–1826. [Google Scholar] [CrossRef]
- Stone, D.; Whalley, L.K.; Heard, D.E. Tropospheric OH and HO2 radicals: Field measurements and model comparisons. Chem. Soc. Rev. 2012, 41, 6348–6404. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; An, Z.; Li, M.; Huo, Y.; Zhou, Y.; Xie, J.; He, M. Comparison of ribavirin degradation in the UV/H2O2 and UV/PDS systems: Reaction mechanism, operational parameter and toxicity evaluation. J. Environ. Chem. Eng. 2023, 11, 109193. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06 class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 119, 215–241. [Google Scholar] [CrossRef] [Green Version]
- de Souza, G.L.; Peterson, K.A. Benchmarking antioxidant-related properties for gallic acid through the use of DFT, MP2, CCSD, and CCSD (T) approaches. J. Phys. Chem. A 2021, 125, 198–208. [Google Scholar] [CrossRef]
- Arathala, P.; Tangtartharakul, C.B.; Sinha, A. Atmospheric ring-closure and dehydration reactions of 1, 4-hydroxycarbonyls in the gas phase: The impact of catalysts. J. Phys. Chem. A 2021, 125, 5963–5975. [Google Scholar] [CrossRef]
- Arathala, P.; Musah, R.A. Theoretical Study of the Atmospheric Chemistry of Methane Sulfonamide Initiated by OH Radicals and the CH3S (O) 2N• H+ 3O2 Reaction. J. Phys. Chem. A 2022, 126, 9447–9460. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Mei, Q.; Wei, B.; An, Z.; Sun, J.; Xie, J.; He, M. Mechanism and kinetics of ClO-mediated degradation of aromatic compounds in aqueous solution: DFT and QSAR studies. Chem. Eng. J. 2021, 412, 128728. [Google Scholar] [CrossRef]
- Sun, J.; Wei, B.; Mei, Q.; An, Z.; Wang, X.; He, M. Ozonation of 3-methylcatechol and 4-methylcatechol in the atmosphere and aqueous particles: Mechanism, kinetics and ecotoxicity assessment. Chem. Eng. J. 2019, 358, 456–466. [Google Scholar] [CrossRef]
- Wang, X.; Sun, J.; Han, D.; Bao, L.; Mei, Q.; Wei, B.; An, Z.; He, M.; Yuan, S.; Xie, J.; et al. Gaseous and heterogeneous reactions of low-molecular-weight (LMW) unsaturated ketones with O3: Mechanisms, kinetics, and effects of mineral dust in tropospheric chemical processes. Chem. Eng. J. 2020, 395, 125083. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Long, B.; Xia, Y.; Truhlar, D.G. Quantitative Kinetics of HO2 Reactions with Aldehydes in the Atmosphere: High-Order Dynamic Correlation, Anharmonicity, and Falloff Effects Are All Important. J. Am. Chem. Soc. 2022, 144, 19910–19920. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Meana-Pañeda, R.; Truhlar, D.G. Prediction of experimentally unavailable product branching ratios for biofuel combustion: The role of anharmonicity in the reaction of isobutanol with OH. J. Am. Chem. Soc. 2014, 136, 5150–5160. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Canneaux, S.; Bohr, F.; Henon, E. KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 2014, 35, 82–93. [Google Scholar] [CrossRef]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Ecosar V 2.0, U.S.E.P.A., Washington, D.C. Available online: https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model (accessed on 8 August 2022).
- T.E.S.T. V 5.1, U.S.E., 2020. User’s Guide for T. E. S. T. (Toxicity Estimation Software Tool) Version 5.1: A Java Application to Estimate Toxicities and Physical Properties from Molecular Structure. Available online: https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test (accessed on 21 October 2022).
- Spielmann, H. White Paper of the Commission of European Community—Strategy for a future chemicals policy. Altex 2001, 18, 147–148. [Google Scholar] [PubMed]
- Arathala, P.; Musah, R.A. Thermochemistry and kinetics of the atmospheric oxidation reactions of propanesulfinyl chloride initiated by OH radicals: A computational approach. J. Phys. Chem. A 2022, 126, 4264–4276. [Google Scholar] [CrossRef] [PubMed]
- Kurylo, M.J.; Orkin, V.L. Determination of atmospheric lifetimes via the measurement of OH radical kinetics. Chem. Rev. 2003, 103, 5049–5076. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Schaefer, T.; He, L.; Zhang, Y.; Sun, X.; Ventura, O.N.; Herrmann, H. T-and pH-dependent kinetics of the reactions of· OH (aq) with glutaric and adipic acid for atmospheric aqueous-phase chemistry. ACS Earth Space Chem. 2021, 5, 1854–1864. [Google Scholar] [CrossRef]
- Truhlar, D.G. Nearly encounter-controlled reactions: The equivalence of the steady-state and diffusional viewpoints. J. Chem. Educ. 1985, 62, 104. [Google Scholar] [CrossRef]
- Steubing, W. Ueber die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in Flüssigkeiten suspendierten Teilchen. Z. Für Chem. Und Ind. Der Kolloide 1908, 3, 549–560. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wei, B.; Tang, R. Theoretical Study on the Mechanisms, Kinetics, and Toxicity Evaluation of OH-Initiated Atmospheric Oxidation Reactions of Coniferyl Alcohol. Atmosphere 2023, 14, 976. https://doi.org/10.3390/atmos14060976
Zhang Y, Wei B, Tang R. Theoretical Study on the Mechanisms, Kinetics, and Toxicity Evaluation of OH-Initiated Atmospheric Oxidation Reactions of Coniferyl Alcohol. Atmosphere. 2023; 14(6):976. https://doi.org/10.3390/atmos14060976
Chicago/Turabian StyleZhang, Yu, Bo Wei, and Rongzhi Tang. 2023. "Theoretical Study on the Mechanisms, Kinetics, and Toxicity Evaluation of OH-Initiated Atmospheric Oxidation Reactions of Coniferyl Alcohol" Atmosphere 14, no. 6: 976. https://doi.org/10.3390/atmos14060976
APA StyleZhang, Y., Wei, B., & Tang, R. (2023). Theoretical Study on the Mechanisms, Kinetics, and Toxicity Evaluation of OH-Initiated Atmospheric Oxidation Reactions of Coniferyl Alcohol. Atmosphere, 14(6), 976. https://doi.org/10.3390/atmos14060976