Are the Fenno-Scandinavian Arctic Wetlands a Significant Regional Source of Formic Acid?


 ,
,  ,
,  and
and
Abstract
:1. Introduction
1.1. Arctic Wetland Source of HC(O)OH
1.2. Campaign Outline
2. Experimental
2.1. CIMS Measurement Technique
2.2. Formic Acid and HCN Calibration
2.3. CH4 Measurements (FGGA and WAS Sampling)
2.4. CO, O3 and Additional Supporting Measurements (FAAM BAe-146 Core Instruments)
3. Flux Calculation
4. Case Studies
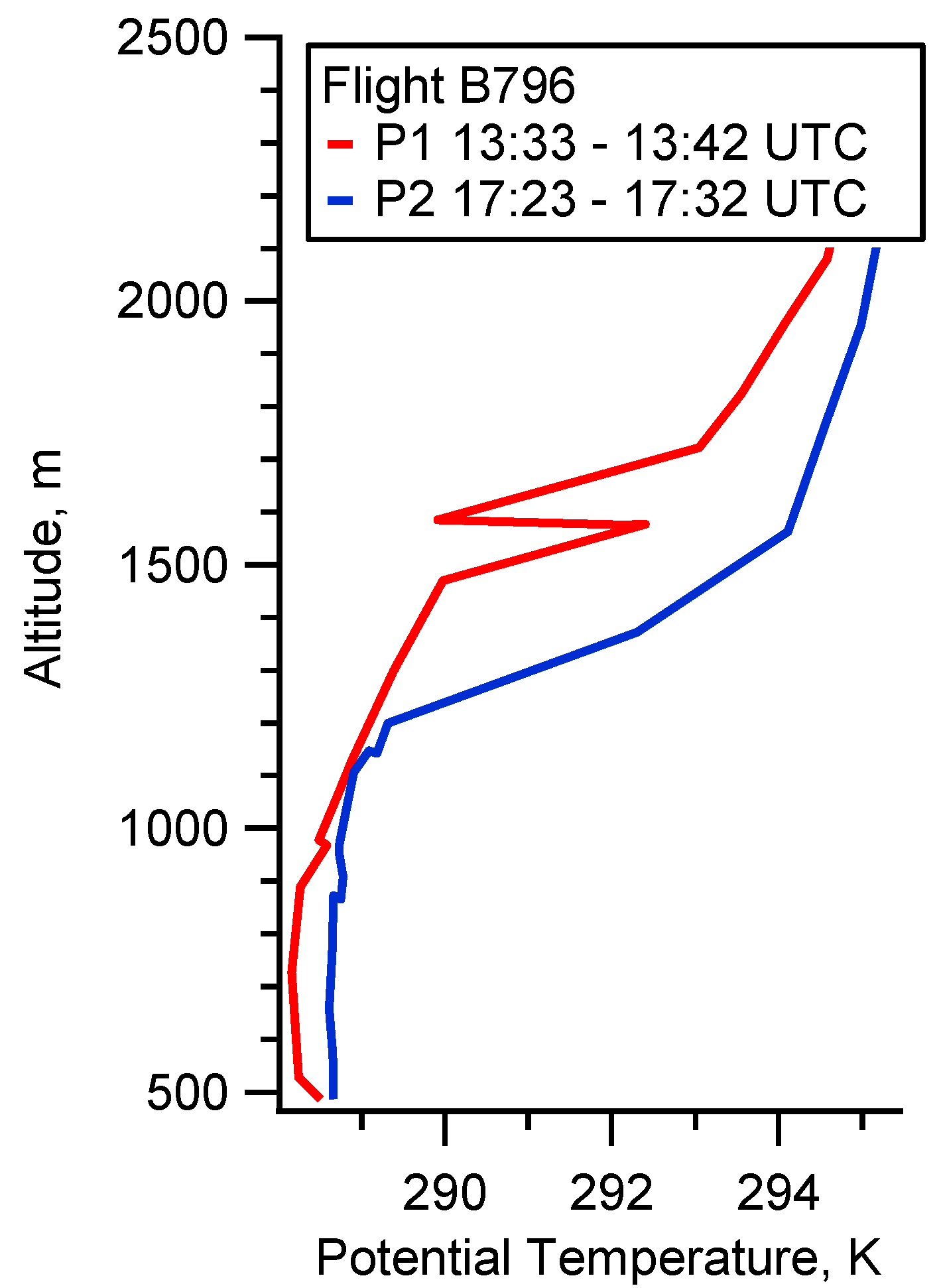
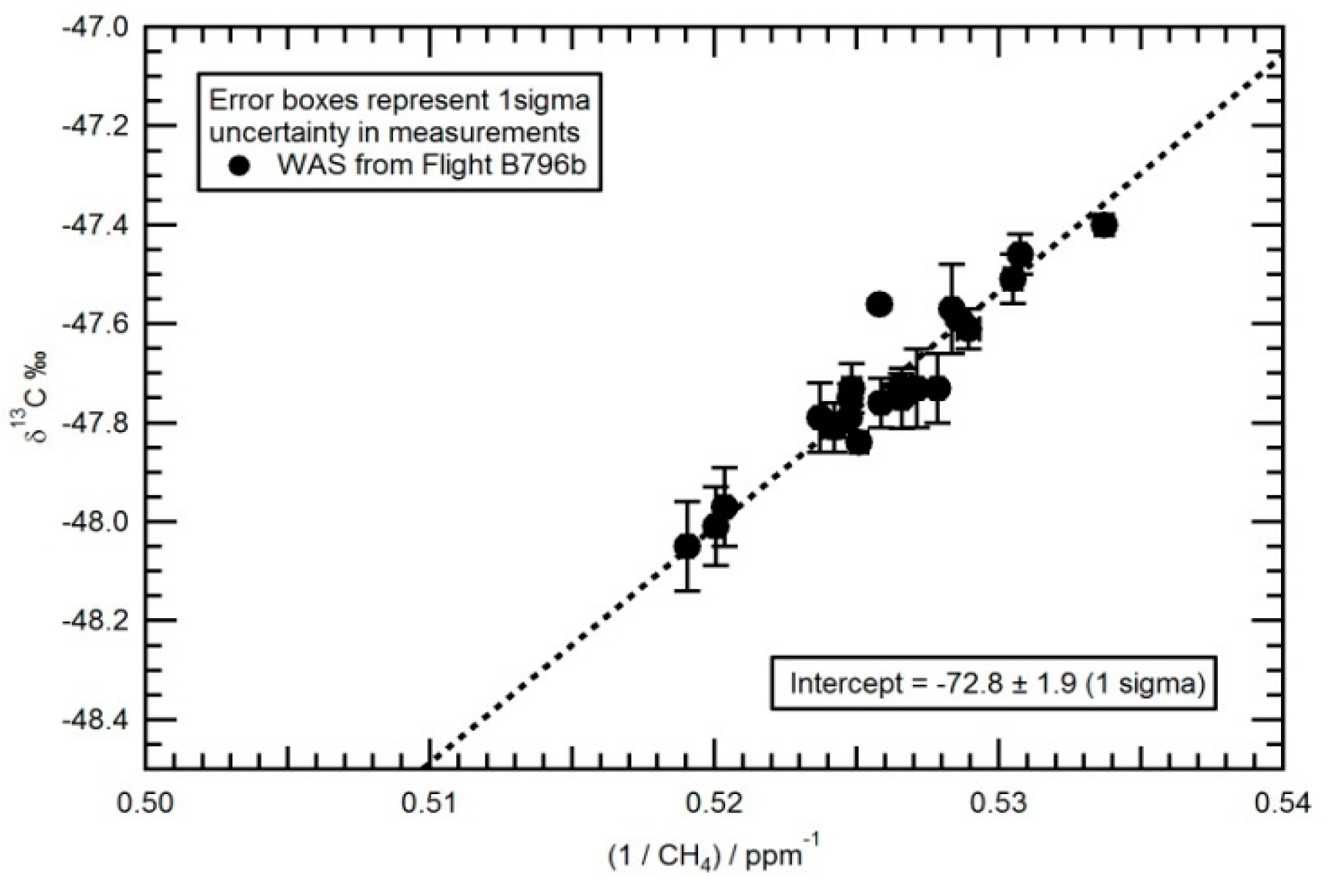
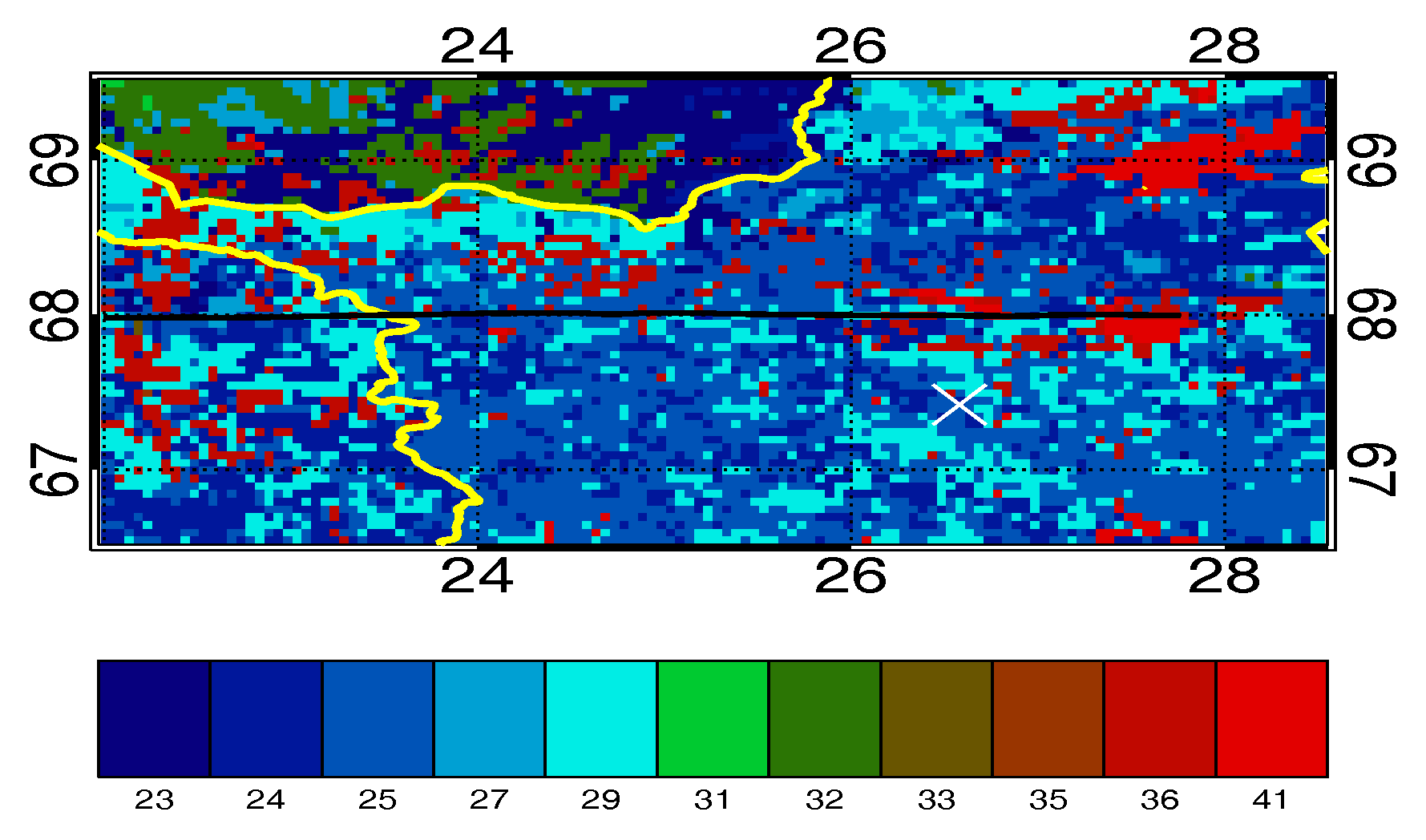
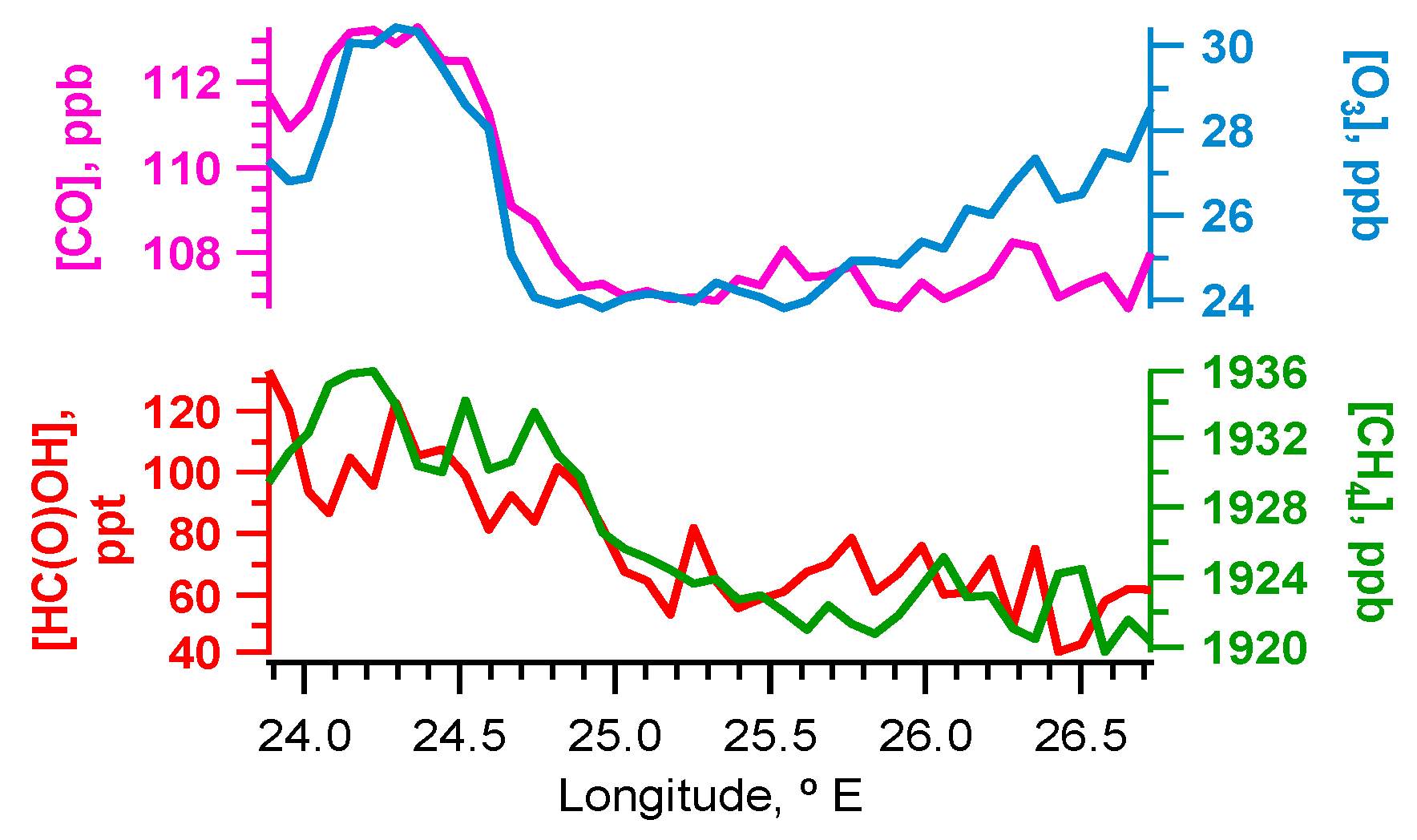
4.1. Case Study B796b
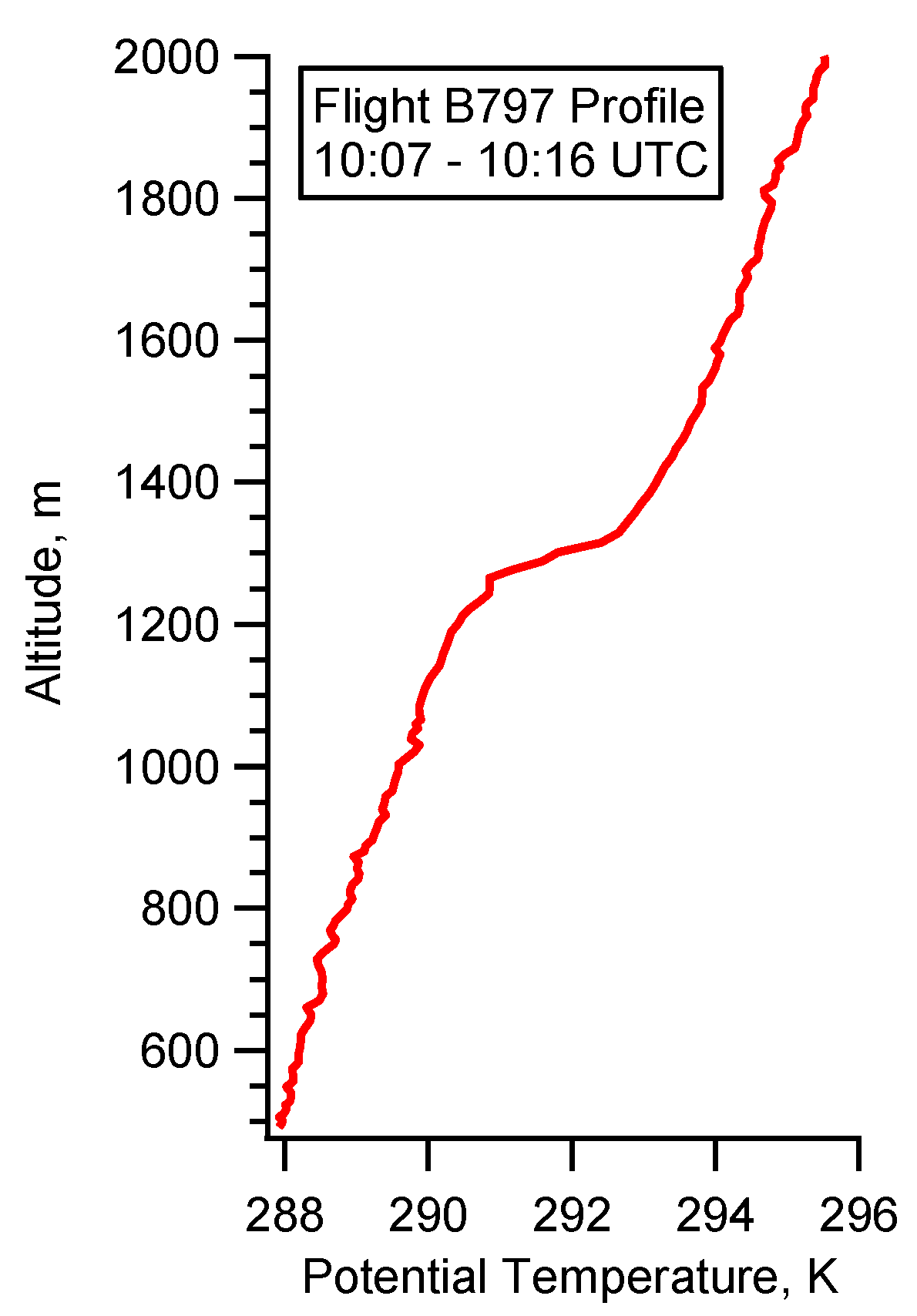
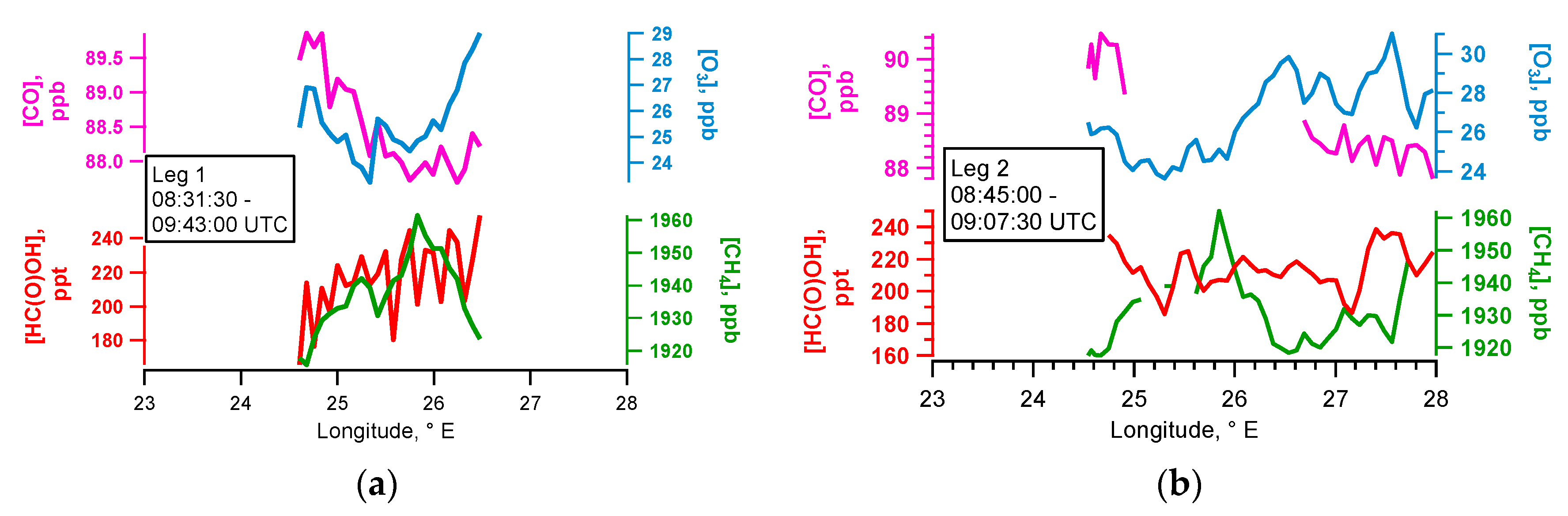
4.2. Case Study B797
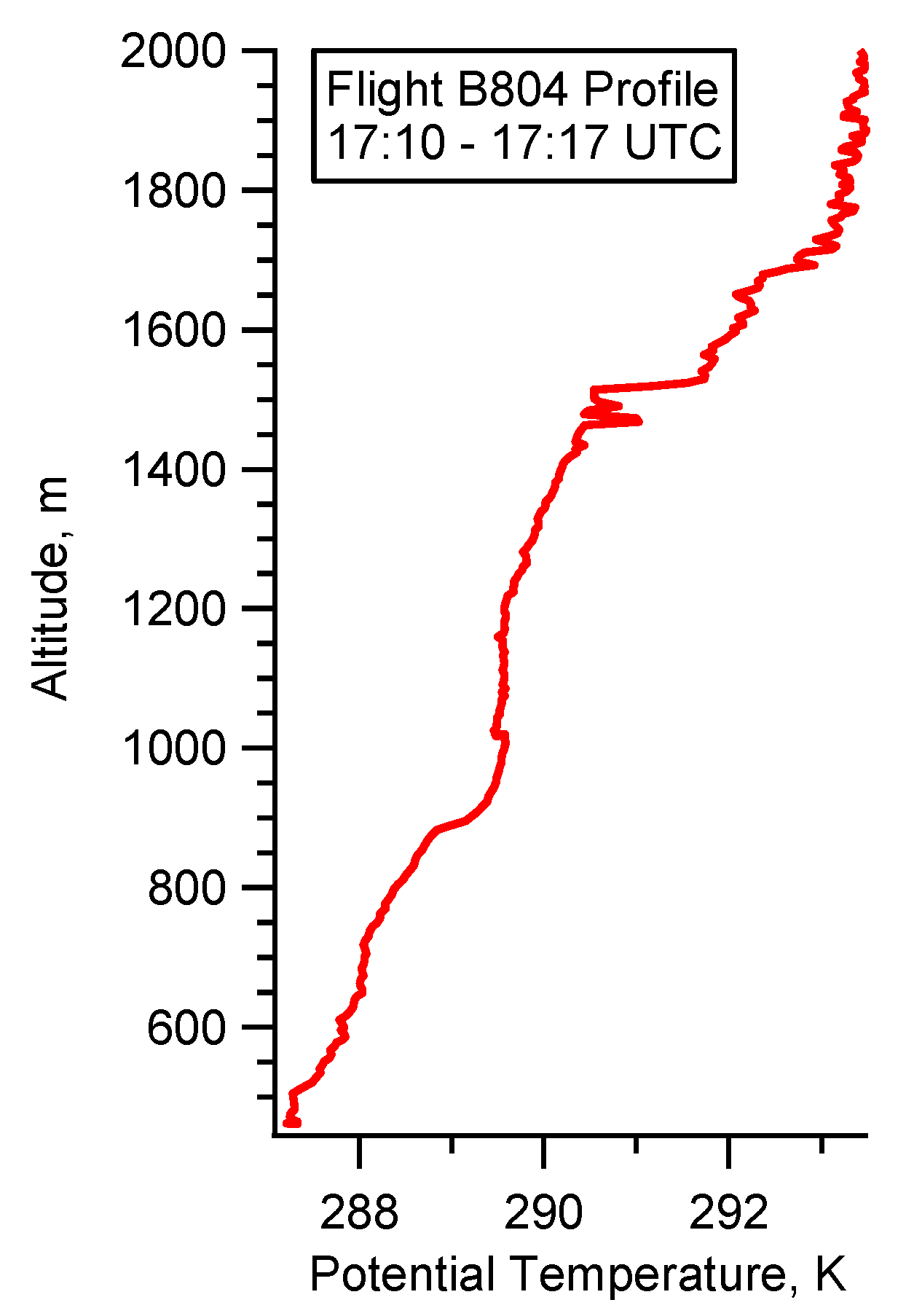
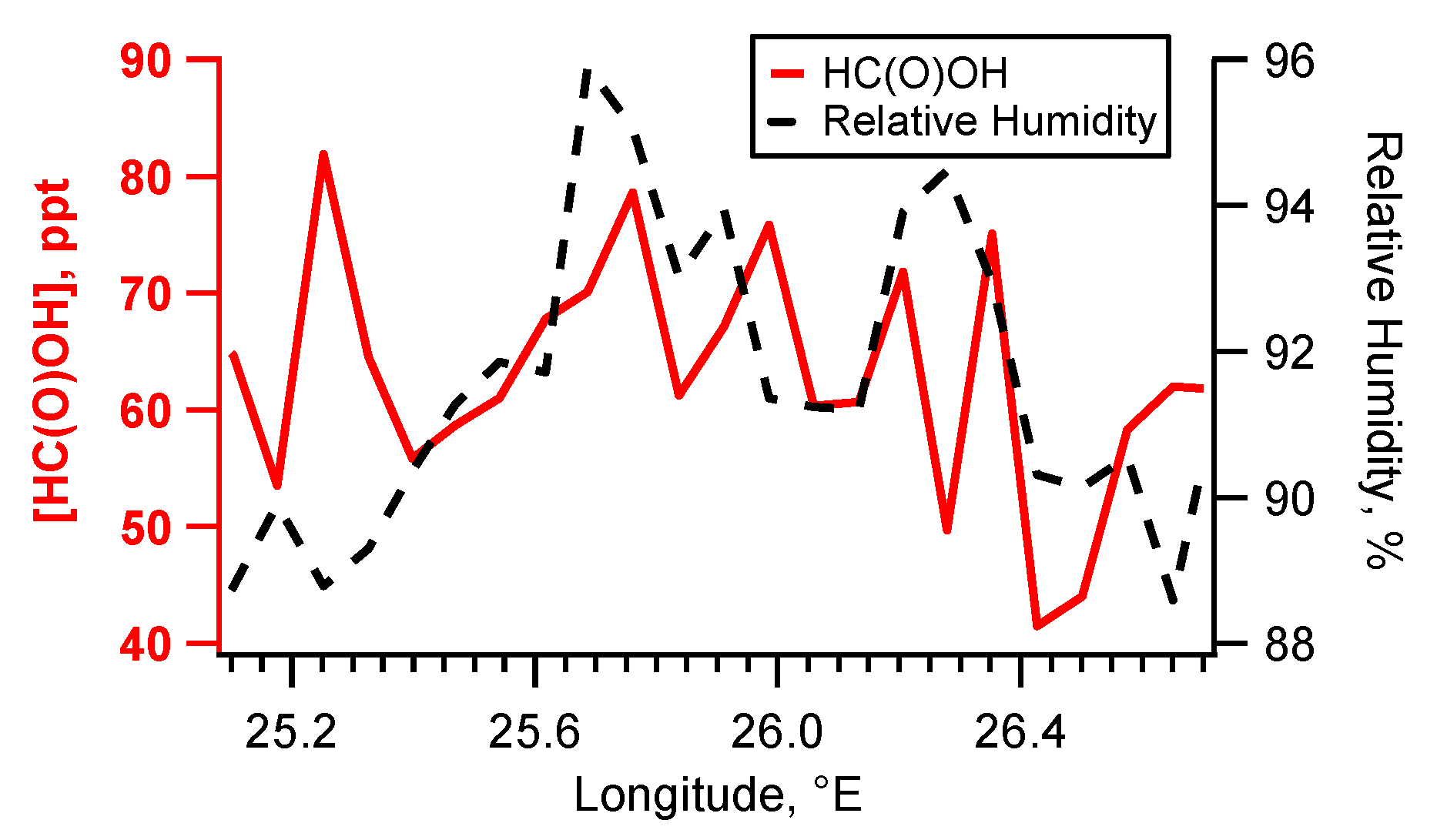
4.3. Case Study B804
5. Results and Discussion
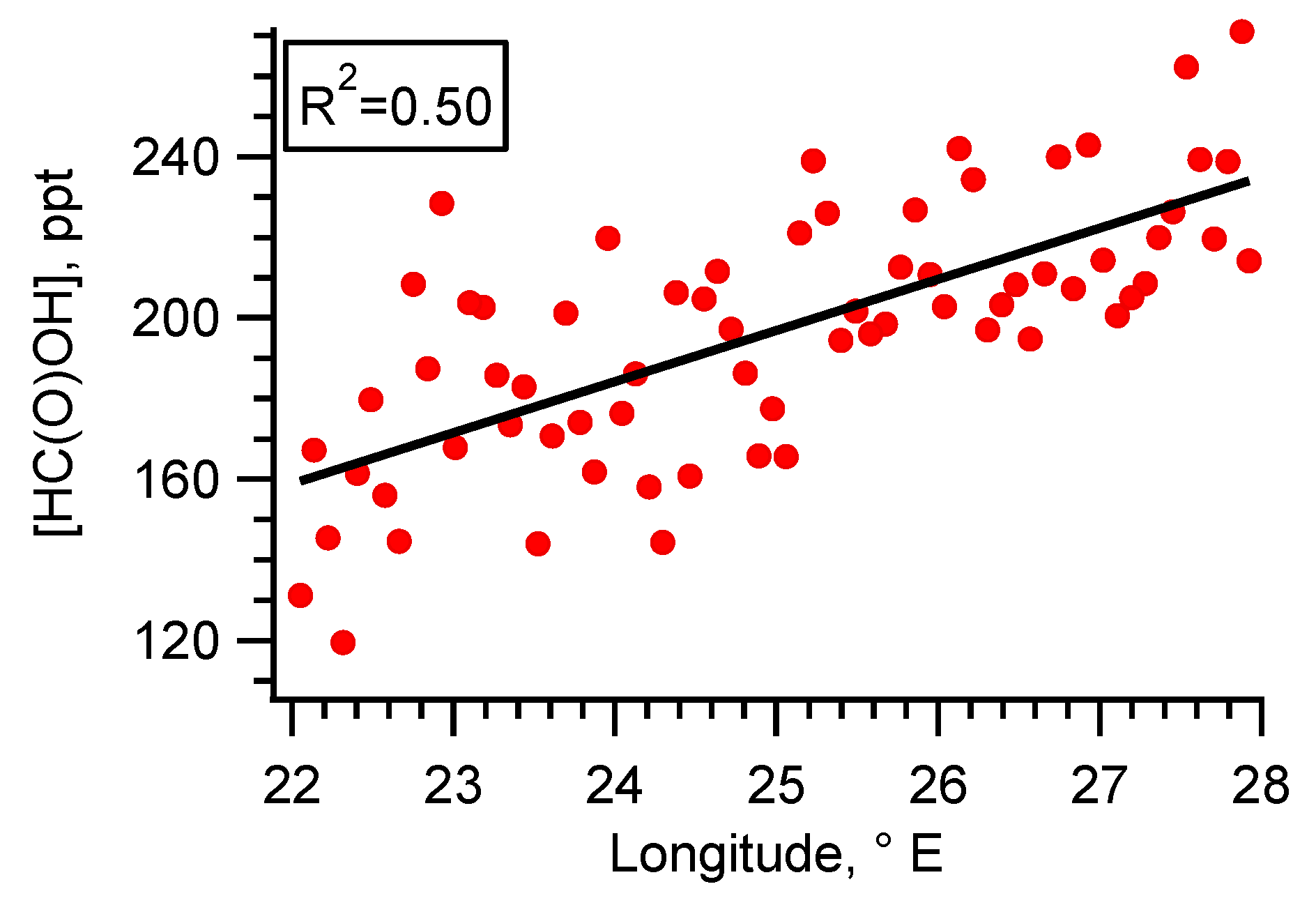
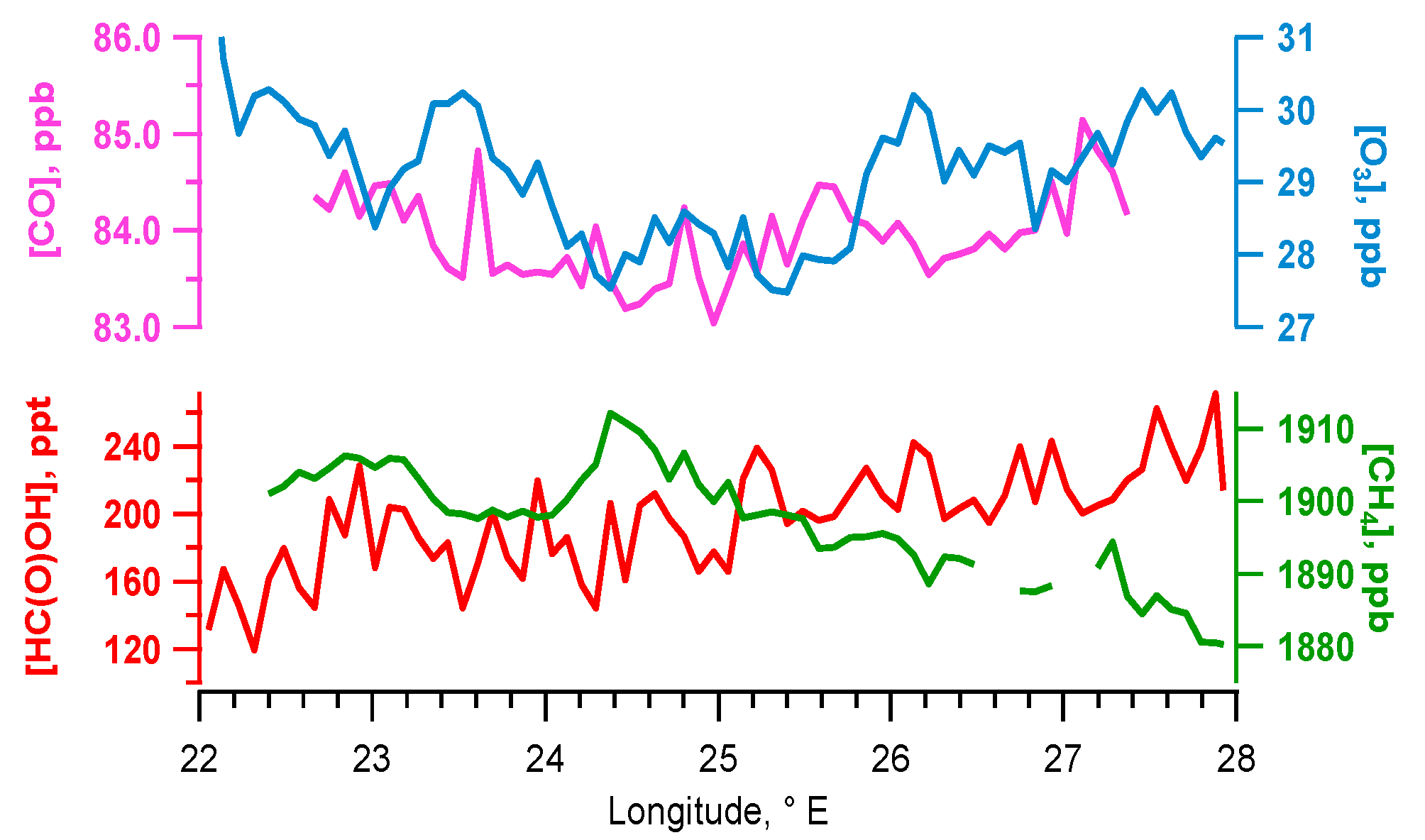
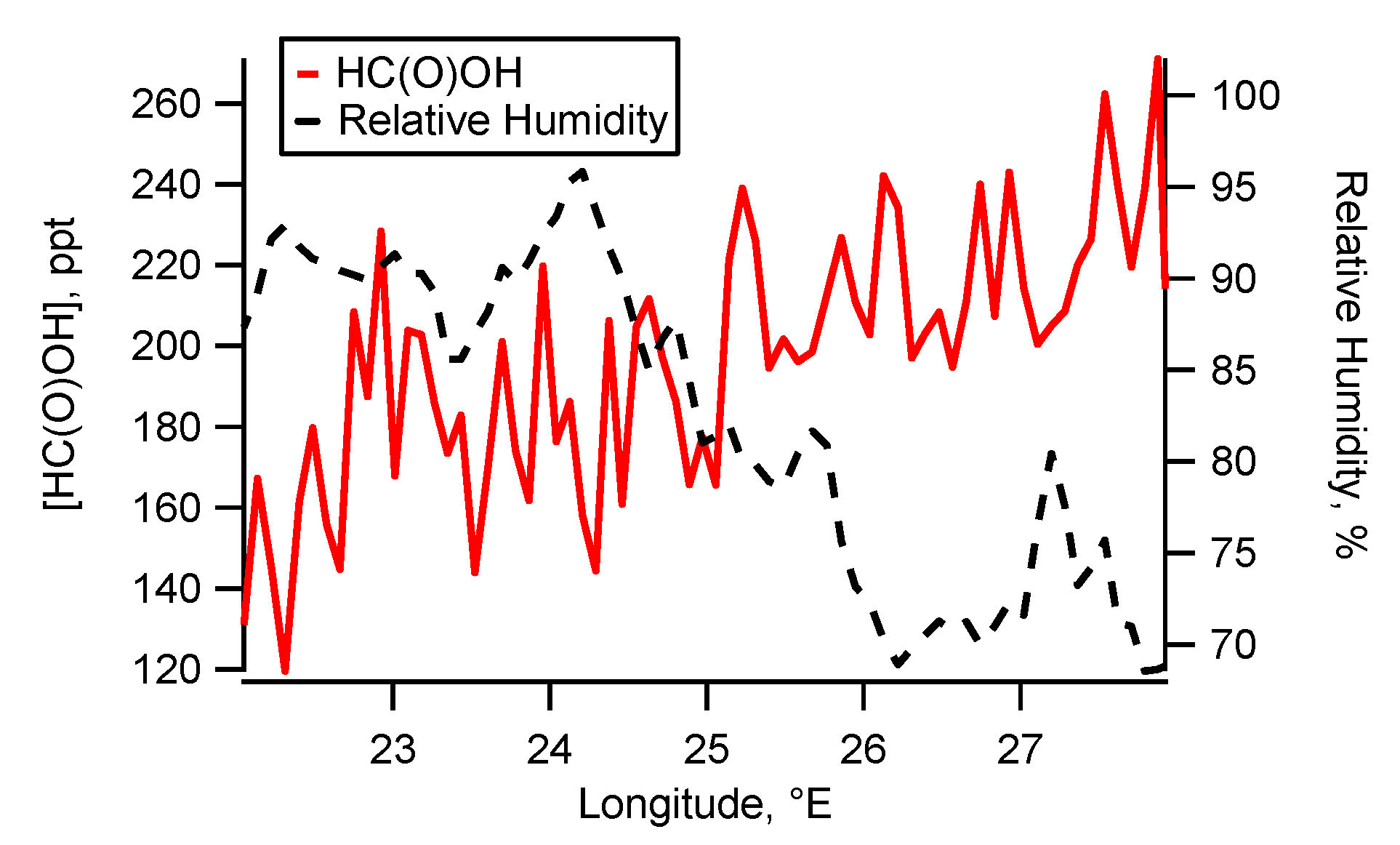
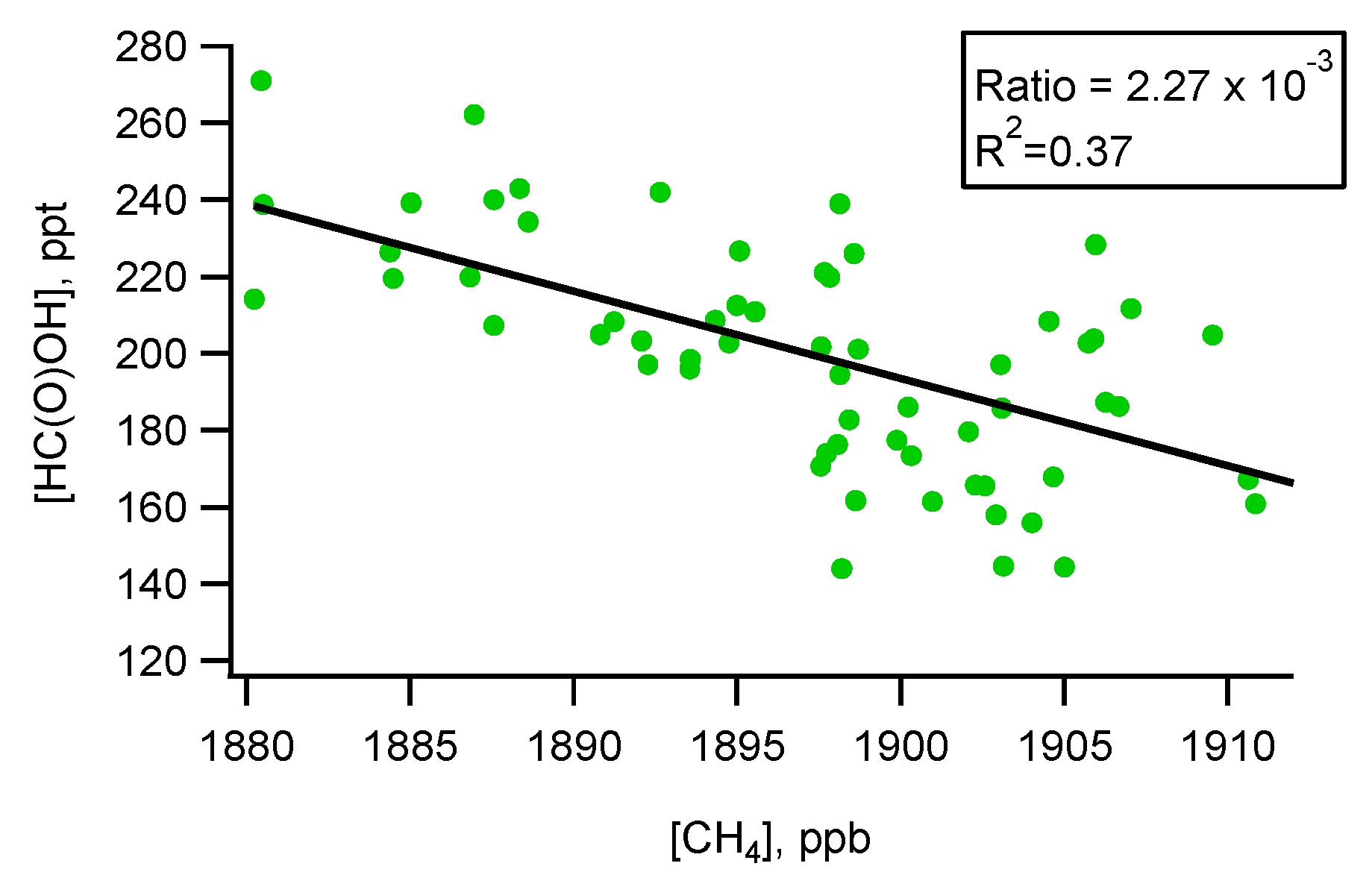
5.1. Flight B796b: HC(O)OH Flux
Source Attribution
5.2. Flight B797: HC(O)OH Flux
5.3. Flight B804: HC(O)OH Flux
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dawson, G.A.; Farmer, J.C.; Jarvis, L.M. Formic and acetic acids in the atmosphere of the southwest U.S.A. Geophys. Res. Lett. 1980, 7, 725–728. [Google Scholar] [CrossRef]
- Keene, W.C.; Galloway, J.N.; Holden, J.D. Measurement of weak organic acidity in precipitation from remote areas of the world. J. Geophys. Res. 1983, 88, 5122–5130. [Google Scholar] [CrossRef]
- Andreae, M.O.; Talbot, R.W.; Andreae, T.W.; Harriss, R.C. Formic and acetic acid over the central amazon region, Brazil 1. Dry season. J. Geophys. Res. 1988, 93, 1616–1624. [Google Scholar] [CrossRef]
- Bannan, T.J.; Bacak, A.; Muller, J.B.A.; Murray, A.M.; Jones, B.T.; Le Breton, M.; Leather, K.E.; Ghalaieny, M.; Xiao, P.; Shallcross, D.E.; et al. Importance of direct anthropogenic emissions of HC(O)OH measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012. Atmos. Environ. 2013, 83, 301–310. [Google Scholar] [CrossRef]
- Paulot, F.; Wunch, D.; Crounse, J.D.; Toon, J.C.; Millet, B.D.; DeCarlo, P.F.; Vigouroux, C.; Deutscher, N.M.; Abad, G.G.; Notholt, J.; et al. Importance of secondary sources in the atmospheric budgets of formic and acetic acids. Atmos. Chem. Phys. 2011, 11, 1989–2013. [Google Scholar] [CrossRef]
- Jacob, D.J. Chemistry of OH in remote cloud and its role in the production of HC(O)OH and peroxymonosulphate. J. Geophys. Res. 1986, 91, 9807–9826. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Bernath, P.F.; Boone, C.D.; McLeod, S.D.; Manney, G.L.; Toon, G.C. Global distribution of upper tropospheric HC(O)OH from the ACE-FTS. Atmos. Chem. Phys. 2009, 9, 8039–8047. [Google Scholar] [CrossRef]
- Stavrakou, T.; Müller, J.-F.; Peeters, J.; Razavi, A.; Clarisse, L.; Clerbaux, C.; Coheur, P.F.; Hurtmans, D.; De Mazière, M.; Vigouroux, C.; et al. Satellite evidence for a large source of HC(O)OH from boreal and tropical forests. Nat. Geosci. 2011, 5, 26–30. [Google Scholar] [CrossRef]
- Le Breton, M.; McGillen, M.R.; Muller, J.B.A.; Bacak, A.; Shallcross, D.E.; Xiao, P.; Huey, L.G.; Tanner, D.J.; Coe, H.; Percival, C.J. Airborne observations of formic acid using a chemical ionisation mass spectrometer. Atmos. Meas. Tech. 2012, 5, 3029–3039. [Google Scholar] [CrossRef]
- Neeb, P.; Sauer, F.; Horie, O.; Moortgat, G.K. Formation of hydroxylmethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of waer vapour. Atmos. Environ. 1997, 31, 1417–1423. [Google Scholar] [CrossRef]
- Kesselmeier, J.; Bode, K.; Gerlach, C.; Jork, E.-M. Exchange of atmospheric formic and acetic acids with trees and crop plants under controlled chamber and purified air conditions. Atmos. Environ. 1998, 32, 1765–1775. [Google Scholar] [CrossRef]
- Sanhueza, E.; Andreae, M.O. Emission of formic and acetic acids from tropical savannah soils. Geophys. Res. Lett. 1991, 18, 1707–1710. [Google Scholar] [CrossRef]
- Kawamura, K.; Ng, L.-L.; Kapian, I.R. Determination of organic acids (C1–C10) in the atmosphere, motor exhausts, and engine oil. Environ. Sci. Technol. 1985, 19, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Talbot, R.W.; Beecher, K.M.; Hariss, R.C.; Cofer, W.R., III. Atmospheric geochemistry of formic and acetic acids in mid-latitude temperate site. J. Geophys. Res. 1998, 93, 1638–1652. [Google Scholar] [CrossRef]
- Talbot, R.W.; Andreae, M.O.; Berresheim, H.; Jacob, D.J.; Beecher, K.M. Sources and sinks of formic, acetic, and pyruvic acids over central Amazonia 2. Wet season. J. Geophys. Res. 1990, 95, 16799–16811. [Google Scholar] [CrossRef]
- Graedel, T.E.; Weschler, C.J. Chemistry within aqueous atmospheric aerosols and raindrops. Rev. Geophys. 1981, 19, 505–539. [Google Scholar] [CrossRef]
- Jones, B.T.; Muller, J.B.A.; O’Shea, S.; Bacak, A.; Le Breton, M.; Bannan, T.J.; Leather, K.E.; Booth, A.M.; Illingworth, S.; Bower, K.; et al. Airborne measurements of HC(O)OH in the European Arctic: A winter-summer comparison. Atmos. Environ. 2014, 99, 556–567. [Google Scholar] [CrossRef]
- Grutter, M.; Glatthor, N.; Stiller, G.P.; Fischer, H.; Grabowski, U.; Hopfner, M.; Kellmann, S.; Linden, A.; von Clarmann, T. Global distribution and variability of HC(O)OH as observed by MIPAS-ENVISAT. J. Geophys. Res. 2010, 115, D10303. [Google Scholar] [CrossRef]
- Sposito, G. The Chemistry of Soils; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Forster, P.; Ramaswamy, V. Changes in atmospheric constituents and in radiative forcing. In Climate Change 2007: The Physical Science Basis; Cambridge University Press: Cambridge, UK, 2007; pp. 129–234. [Google Scholar]
- Parmentier, F.-J.W.; Christensen, T.R.; Sorensen, L.L.; Rysgaard, S.; McGuire, A.D.; Miller, P.A.; Walker, D.A. The impact of lower sea-ice extent on Arctic greenhouse gas exchange. Nat. Clim. Chang. 2013, 3, 195–202. [Google Scholar] [CrossRef]
- Leibowitz, S.G. Isolated wetlands and their functions: An ecological perspective. Soc. Wetl. Sci. 2003, 23, 517–531. [Google Scholar] [CrossRef]
- Oelke, C.; Zhang, T.J.; Serreze, M.C. Modelling evidence for recent warming of the Arctic soil thermal regime. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Mausbach, M.J.; Richardson, J.L. Biogeochemical processes in hydric soil formation. Curr. Top. Wetl. Biogeochem. 1994, 1, 68–127. [Google Scholar]
- Soil Grids. Available online: https://www.soilgrids.org/#/?lon=22.12646484375&lat=68.29987352961231&zoom=9&layer=geonode:taxnwrb_250m&showInfo=1 (accessed on 14 June 2017).
- Nowak, J.B.; Neuman, J.A.; Kozai, K.; Huey, L.G.; Tanner, D.J.; Holloway, J.S.; Ryerson, T.B.; Frost, G.J.; McKeen, S.A.; Fehsenfeld, F.C. A chemical ionization mass spectrometry technique for airborne measurements of ammonia. J. Geophys. Res. Atmos. 2007, 112, D10S02. [Google Scholar] [CrossRef]
- Slusher, D.L.; Huey, L.G.; Tanner, D.J.; Flocke, F.M.; Roberts, J.M. A thermal dissociation-chemical ionization mass spectrometry (td-cims) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide. J. Geophys. Res. 2004, 109, D19315. [Google Scholar] [CrossRef]
- O’Shea, S.J.; Bauguitte, S.J.B.; Gallagher, M.W.; Lowry, D.; Percival, C.J. Development of a cavity-enhanced absorption spectrometer for airborne measurements of CH4 and CO2. Atmos. Meas. Tech. 2013, 6, 1095–1109. [Google Scholar] [CrossRef]
- Lewis, A.C.; Evans, M.J.; Hopkins, J.R.; Punjabi, S.; Read, K.A.; Purvis, R.M.; Andrews, S.J.; Moller, S.J.; Carpenter, L.J.; Lee, J.D.; et al. The influence of biomass burning on the global distribution of selected non-methane organic compounds. Atmos. Chem. Phys. 2011, 13, 851–867. [Google Scholar] [CrossRef]
- Keeling, C.D. The concentration and isotopic abundance of atmospheric carbon dioxide in rural areas. Geochim. Cosmochim. Acta 1958, 13, 322–334. [Google Scholar] [CrossRef]
- Keeling, C.D. The concentration and isotopic abundances of carbon dioxide in rural and marine air. Geochim. Cosmochim. Acta 1961, 24, 277–298. [Google Scholar] [CrossRef]
- Gerbig, C.; Schmitgen, S.; Kley, D.; Volz-Thomas, A. An improved fast-response vacuum-UV resonance fluorescence CO instrument. J. Geophys. Res. 1999, 104, 1699–1704. [Google Scholar] [CrossRef]
- Real, E.; Law, K.S.; Weinzierl, B.; Fiebig, M.; Petzold, A.; Wild, O.; Methven, J.; Arnold, S.; Stohl, A.; Huntrieser, H.; et al. Processes influencing ozone levels in Alaskan forest fire plumes during long range transport over the North Atlantic. J. Geophys. Res. Atmos. 2007, 112, D10S41. [Google Scholar] [CrossRef]
- O’Shea, S.J.; Allen, G.; Gallagher, M.W.; Bower, K.; Illingworth, S.M.; Muller, J.B.A.; Jones, B.T.; Percival, C.J.; Bauguitte, S.J.-B.; Cain, M.; et al. Methane and carbon dioxide fluxes and their regional scalability for the European Arctic wetlands during the MAMM project in summer 2012. Atmos. Chem. Phys. 2014, 14, 13159–13174. [Google Scholar] [CrossRef]
- White, W.H.; Anderson, J.A.; Blumenthal, D.L.; Husar, R.B.; Gillani, N.V.; Husar, J.D.; Wilson, W.E. Formation and transport of secondary air-pollutants-ozone and aerosols in St-Louise urban plume. Science 1976, 194, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.W.; Choularton, T.W.; Bower, K.N.; Stromberg, I.M.; Beswick, K.M.; Fowler, D.; Hargreaves, K.J. Measurements of methane fluxes on the landscape scale from a wetland area in north Scotland. Atmos. Environ. 1994, 28, 2421–2430. [Google Scholar] [CrossRef]
- Choularton, T.W.; Gallagher, M.W.; Bower, K.N.; Fowler, D.; Zahniser, M.; Kaye, A. Trace gas flux measurements at the landscape scale using boundary-layer budgets. Phil. Trans. R. Soc. A 1995, 351, 357–368. [Google Scholar] [CrossRef]
- Wratt, D.S.; Gimson, N.R.; Brailsford, G.W.; Lassey, K.R.; Bromley, A.M.; Bell, M.J. Estimating regional methane emissions from agriculture using aircraft measurements of concentration profiles. Atmos. Environ. 2001, 35, 497–508. [Google Scholar] [CrossRef]
- Mays, K.L.; Shepson, P.B.; Stirm, B.H.; Karion, A.; Sweeney, C.; Gurney, K.R. Aircraft-based measurements of the carbon footprint of Indianapolis. Environ. Sci. Technol. 2009, 43, 7816–7823. [Google Scholar] [CrossRef] [PubMed]
- Karion, A.; Sweeney, C.; Petron, G.; Frost, G.; Hardesty, R.M.; Kofler, J.; Miller, B.R.; Newberger, T.; Wolter, S.; Banta, R.; et al. Methane emissions estimate from airborne measurements over a western United States natural gas field. Geophys. Res. Lett. 2013, 40, 1–5. [Google Scholar] [CrossRef]
- Le Breton, M.; Bacak, A.; Muller, J.B.A.; O’Shea, S.J.; Xiao, P.; Ashfold, M.N.R.; Cooke, M.C.; Batt, R.; Shallcross, D.E.; Oram, D.E.; et al. Airborne hydrogen cyanide measurements using a chemical ionisation mass spectrometer for the plume identification of biomass burning forest fires. Atmos. Chem. Phys. 2013, 13, 9217–9232. [Google Scholar]
- Veres, P.; Roberts, J.M.; Warneke, C.; Welsh-Bon, D.; Zahniser, M.; Herndon, S.; Fall, R.; de Gouw, J. Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for measurement of gas-phase organic acids in the atmosphere. Int. J. Mass Spectrom. 2008, 274, 48–55. [Google Scholar] [CrossRef]
- Bridgham, S.D.; Cadillo-Quiroz, H.; Keller, J.K.; Zhuang, G. Methane emissions from wetlands: Biogeochemical, microbial, and modelling perspectives from local to global scales. Glob. Chang. Biol. 2013, 19, 1325–1346. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.E.; Sriskantharajah, S.; Lowry, D.; Lanoiselle, M.; Fowler, C.M.R.; James, R.H.; Hermansen, O.; Lund Myhre, C.; Stohl, A.; Greinert, J.; et al. Arctic methane sources: Isoptopic evidence for atmospheric inputs. Geophys. Res. Lett. 2011, 38, L21803. [Google Scholar] [CrossRef]
- Sriskantharajah, S.; Fisher, R.E.; Lowry, D.; Aalto, T.; Hatakka, J.; Aurela, M.; Laurila, T.; Lohila, A.; Kuitunen, E.; Nisbet, E.G. Stable carbon isotope signatures of methane from a Finnish subarctic wetland. Tellus B 2012, 64, 18818. [Google Scholar] [CrossRef]
- Papen, H.; Rennenberg, H. Microbial processes involved in emissions of radiatively important trace gases. In Proceedings of the Transactions 14th International Congress of Soil Sceince, Kyoto, Japan, 12–18 August 1990; Volume II, pp. 232–237. [Google Scholar]
- O’Connor, F.M.; Boucher, O.; Gedney, N.; Jones, C.D.; Folberth, G.A.; Coppell, R.; Friedlingstein, P.; Collins, W.J.; Chappellaz, J.; Ridley, J.; et al. Possible role of wetlands, permafrost, and methane hydrates in the methane cycle under future climate change: A review. Revs. Geophys. 2010, 48, RG4005. [Google Scholar] [CrossRef]
- Corine Land Cover 2006. Available online: http://www.eea.europa.eu/data-and-maps/data/corine-land-cover-2006-raster (accessed on 22 June 2017).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | B796b | B797 (Combined) | B804 | |||
|---|---|---|---|---|---|---|
| Mean Value | Variability (1σ) | Mean Value | Variability (1σ) | Mean Value | Variability (1σ) | |
| Wind Speed (U) | 2.9 m·s−1 | ±0.9 m·s−1 | 5.9 m·s−1 | ±0.6 m·s−1 | 9.3 m·s−1 | ±1.1 m·s−1 |
| Wind direction | 315.6° | ±26.5° | 342.4° | ±13.5° | 309.9° | ±5.6° |
| Alignment difference between flight track and wind component (cosθ) | 0.731 | 0.315 | 0.280 | 0.227 | 0.796b | 0.076 |
| [HC(O)OH] enhancement over transect x from background | 143 ppt | ±30 ppt | 51 ppt | ±12 ppt | −40 ppt | ±10 ppt |
| HC(O)OH enhancement vs transect x longitude (R2 fit) | 0.51 | 0.03 | 0.08 | |||
| Boundary Layer Height (Z1) | 1330 m | ±130 m | 1310 m | ±100 m | 1475 m | ±100 m |
| Total Flux (mg·m−2·h−1) | 0.0098 | ±0.0057 | 0.0096 | ±0.0079 | −0.0146 | ±0.0029 |
| Number | Land Type | Surface-Type Proportion Occupying Transect x in the Flight Track for B796b, % |
|---|---|---|
| 18 * | Pastures | 0.02 |
| 21 * | Agricultural land with significant natural vegetation | 0.05 |
| 23 | Broad-leaf forest | 5.1 |
| 24 | Coniferous forest | 26.9 |
| 25 | Mixed forest | 22.2 |
| 27 | Moors and heathland | 1.8 |
| 29 | Woodland shrub | 12.2 |
| 31 | Bare rocks | 0 |
| 32 | Sparsely vegetated areas | 0 |
| 33 | Burnt areas | 0 |
| 35 * | Inland marshes | 0.002 |
| 36 | Peat bogs | 24.3 |
| 40 * | Water courses | 0.3 |
| 41 | Water bodies | 7.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, B.T.; Muller, J.; O’Shea, S.; Bacak, A.; Allen, G.; Gallagher, M.; Bower, K.; Le Breton, M.; Bannan, T.J.; Bauguitte, S.; et al. Are the Fenno-Scandinavian Arctic Wetlands a Significant Regional Source of Formic Acid? Atmosphere 2017, 8, 112. https://doi.org/10.3390/atmos8070112
Jones BT, Muller J, O’Shea S, Bacak A, Allen G, Gallagher M, Bower K, Le Breton M, Bannan TJ, Bauguitte S, et al. Are the Fenno-Scandinavian Arctic Wetlands a Significant Regional Source of Formic Acid? Atmosphere. 2017; 8(7):112. https://doi.org/10.3390/atmos8070112
Chicago/Turabian StyleJones, Benjamin T., Jennifer Muller, Sebastian O’Shea, Asan Bacak, Grant Allen, Martin Gallagher, Keith Bower, Michael Le Breton, Thomas J. Bannan, Stephane Bauguitte, and et al. 2017. "Are the Fenno-Scandinavian Arctic Wetlands a Significant Regional Source of Formic Acid?" Atmosphere 8, no. 7: 112. https://doi.org/10.3390/atmos8070112
APA StyleJones, B. T., Muller, J., O’Shea, S., Bacak, A., Allen, G., Gallagher, M., Bower, K., Le Breton, M., Bannan, T. J., Bauguitte, S., Pyle, J., Lowry, D., Fisher, R., France, J., Nisbet, E., Shallcross, D., & Percival, C. (2017). Are the Fenno-Scandinavian Arctic Wetlands a Significant Regional Source of Formic Acid? Atmosphere, 8(7), 112. https://doi.org/10.3390/atmos8070112