Expanded Application of the Passive Flux Meter: In-Situ Measurements of 1,4-Dioxane, Sulfate, Cr(VI) and RDX


Abstract
:1. Introduction
2. Material and Methods
2.1. Passive Flux Meter Technology
2.2. Sorbent Materials
2.3. Extraction and Analytical Procedures
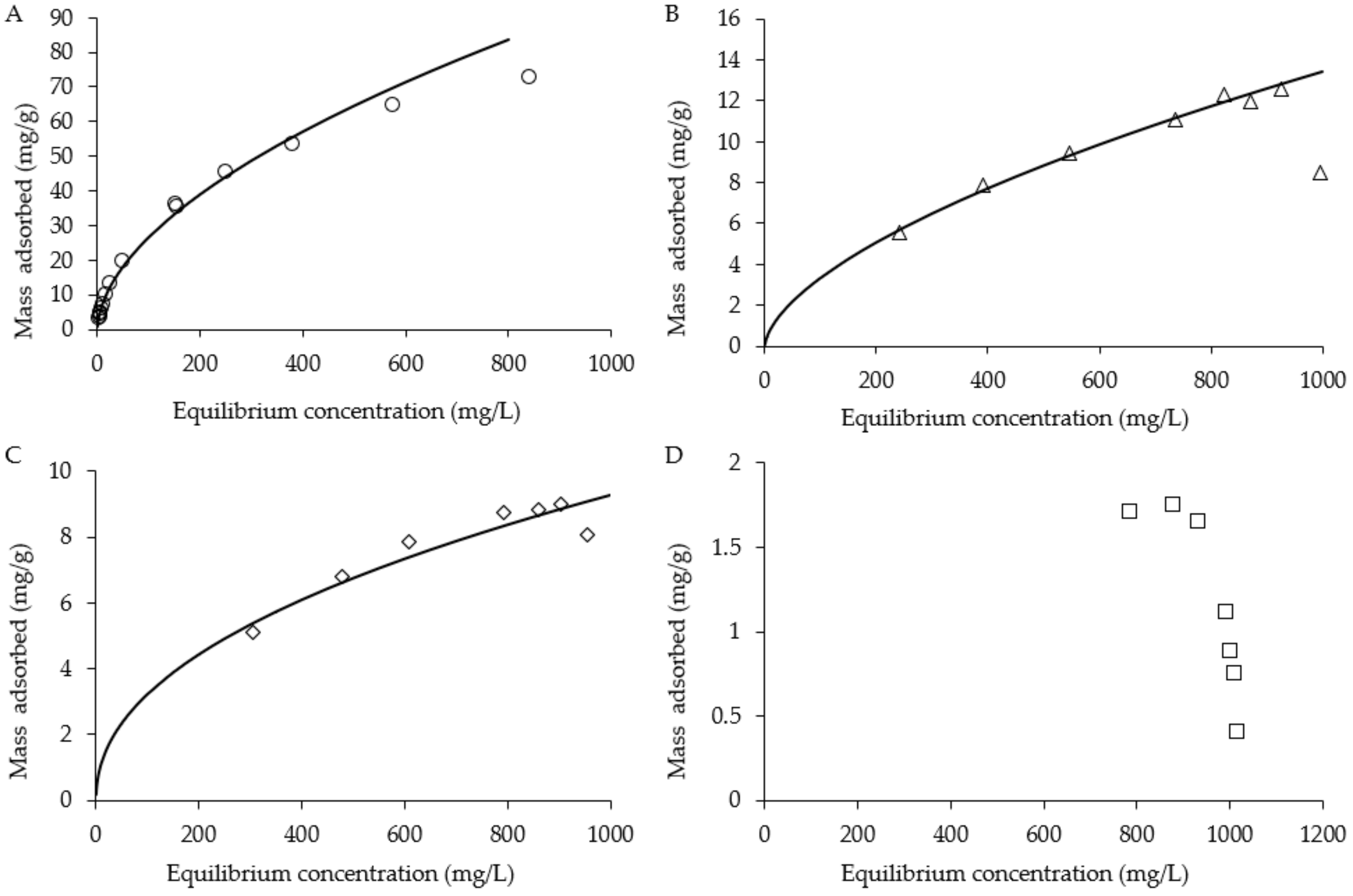
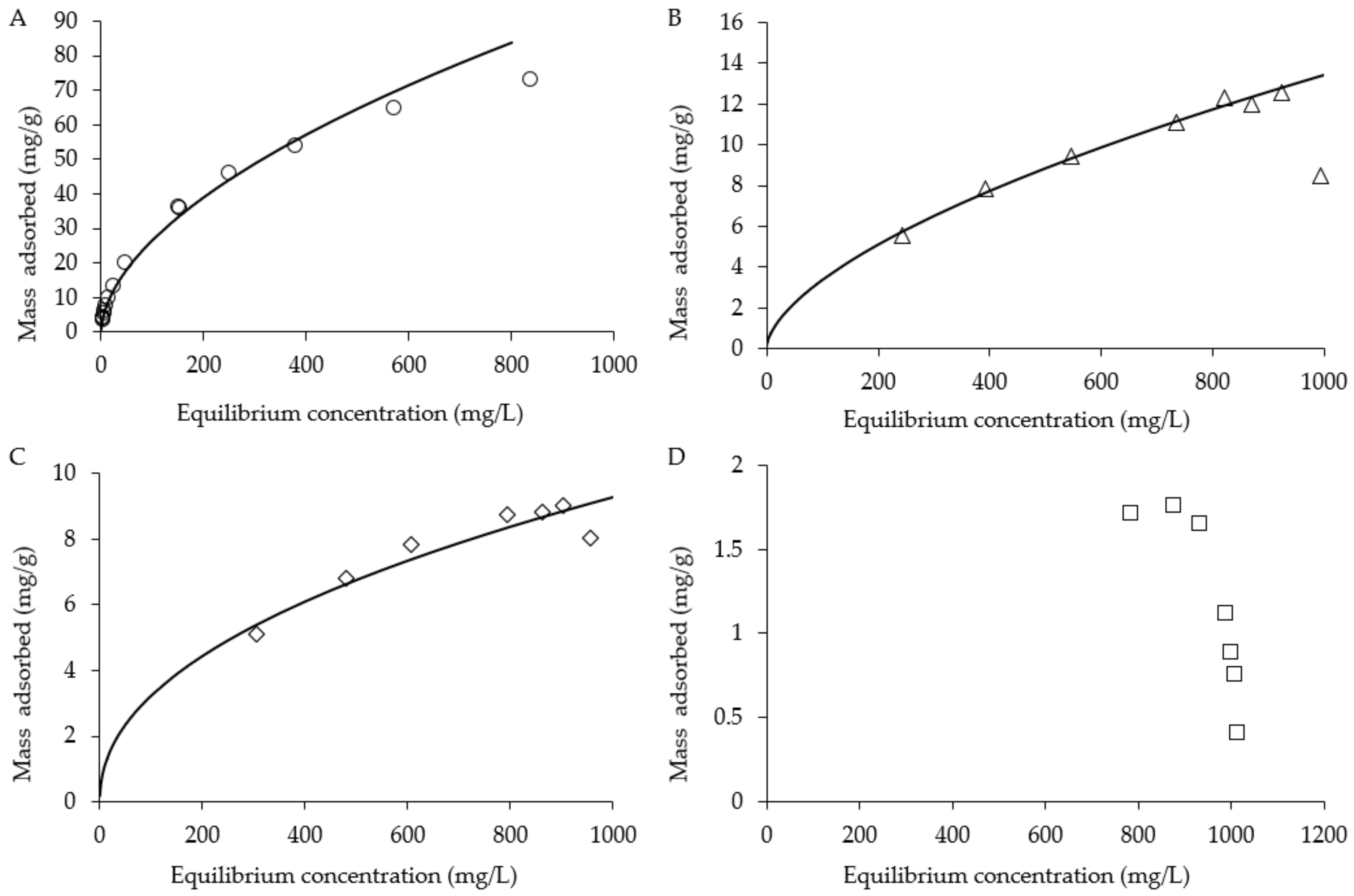
2.4. Batch Adsorption Isotherms and Recovery
2.5. Column Studies
2.6. Bench-Scale Aquifer Testing of 1,4-Dioxane PFM
2.7. Field Studies
2.7.1. RDX and Cr(VI) Measurements
2.7.2. 1,4-Dioxane Measurements
2.7.3. Sulfate Measurements
3. Results
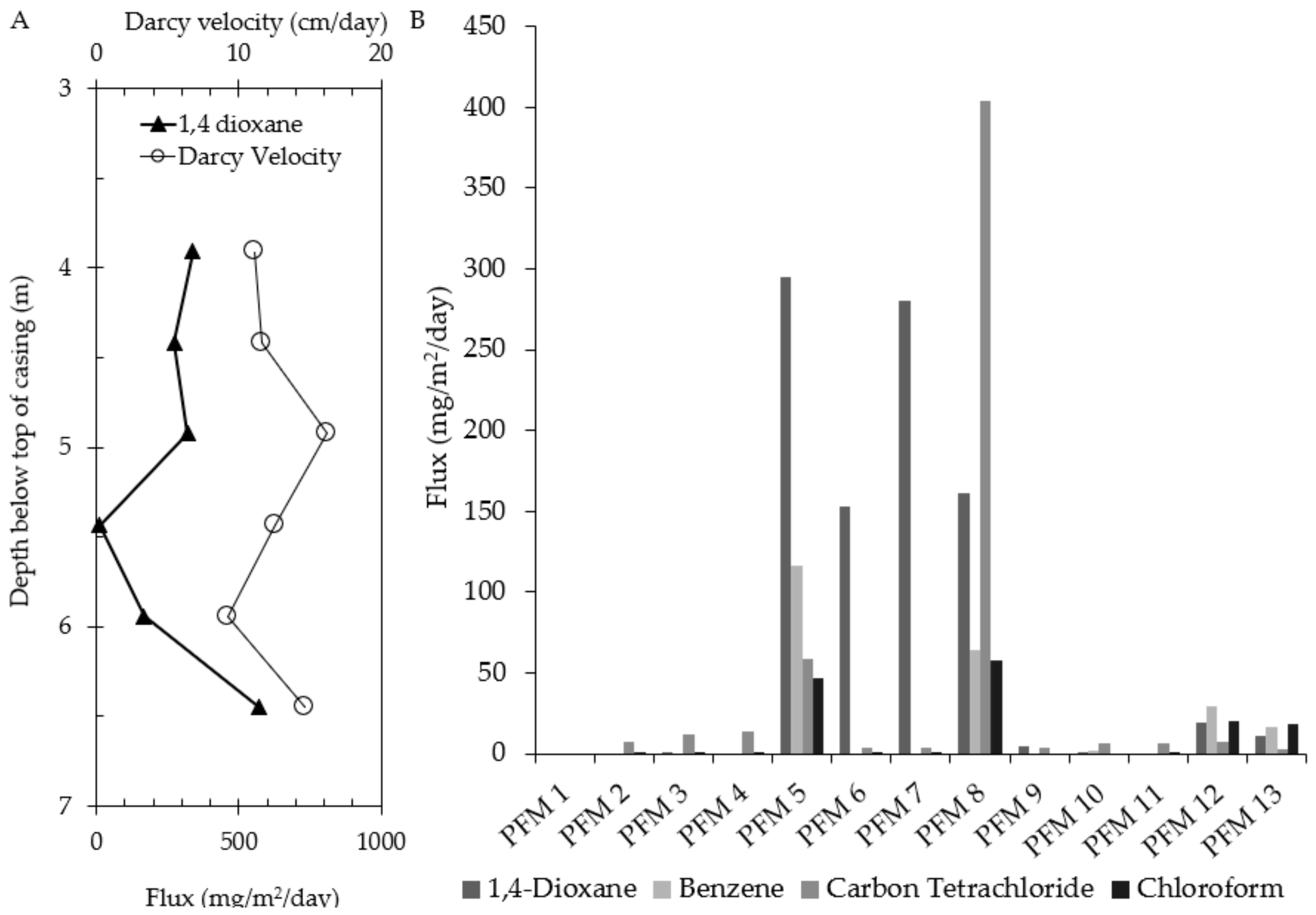
3.1. RDX and Cr(VI)
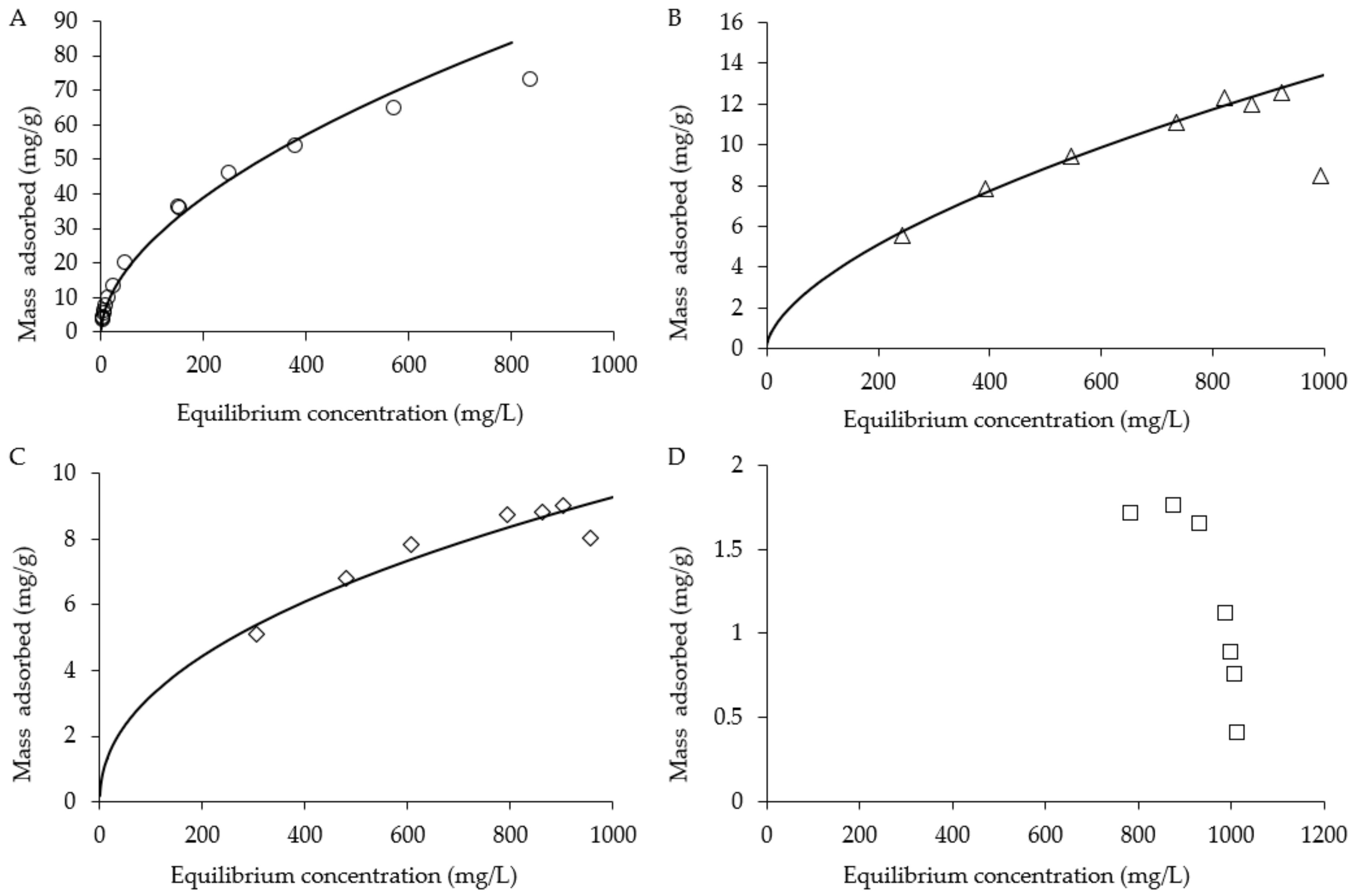
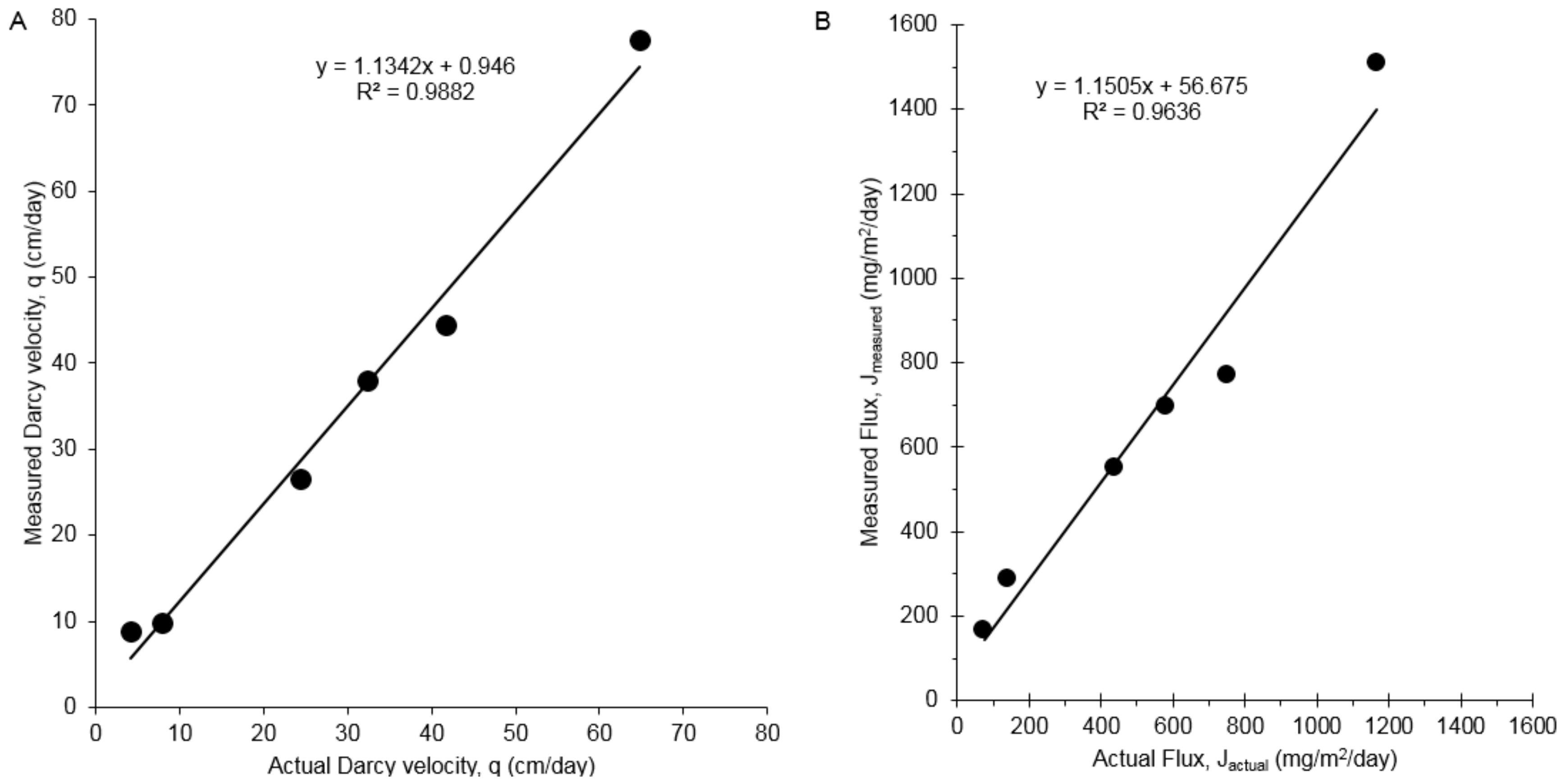
3.2. 1,4-Dioxane
3.3. In-situ Biogenic SO42− Reduction Rates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rao, P.S.C.; Jawitz, J.W.; Enfield, C.G.; Falta, R.W.; Annable, M.D.; Wood, A.L. Technology Integration for Contaminated Site Remediation: Clean-Up Goals and Performance Criteria. In Groundwater Quality: Natural and Enhanced Restoration of Groundwater Pollution; International Association of Hydrological Sciences: Sheffield, UK, 2002; Volume 571–578, Available online: http://hydrologie.org/redbooks/a275/iahs_275_571.pdf (accessed on 16 September 2018).
- Verreydt, G.; Bronders, J.; Van Keer, I.; Diels, L.; Vanderauwera, P. Passive samplers for monitoring vocs in groundwater and the prospects related to mass flux measurements. Ground Water Monit. Remed. 2010, 30, 114–126. [Google Scholar] [CrossRef]
- Martin, H.; Piepenbrink, M.; Grathwohl, P. Ceramic dosimeters for time-integrated contaminant monitoring. J Process Anal. Chem. 2001, 6, 68–73. [Google Scholar]
- Vroblesky, D.A.; Campbell, T.R. Equilibration times, compound selectivity, and stability of diffusion samplers for collection of ground-water VOC concentrations. Adv. Environ. Res. 2001, 5, 1–12. [Google Scholar] [CrossRef]
- Harter, T.; Talozi, S. Evaluation of a simple, inexpensive dialysis sampler for small diameter monitoring wells. Ground Water Monit. Remed. 2004, 24, 97–105. [Google Scholar] [CrossRef]
- Jalalizadeh, M.; Ghosh, U. In situ passive sampling of sediment porewater enhanced by periodic vibration. Environ. Sci. Technol. 2016, 50, 8741–8749. [Google Scholar] [CrossRef] [PubMed]
- Webster, I.T.; Teasdale, P.R.; Grigg, N.J. Theoretical and experimental analysis of peeper equilibration dynamics. Environ. Sci. Technol. 1998, 32, 1727–1733. [Google Scholar] [CrossRef]
- Vrana, B.; Mills, G.A.; Dominiak, E.; Greenwood, R. Calibration of the chemcatcher passive sampler for the monitoring of priority organic pollutants in water. Environ. Pollut. 2006, 142, 333–343. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.; Rothenberg, G. New device and method for flux-proportional sampling of mobile solutes in soil and groundwater. Environ. Sci. Technol. 2005, 39, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Jalalizadeh, M.; Ghosh, U. Analysis of measurement errors in passive sampling of porewater PCB concentrations under static and periodically vibrated conditions. Environ. Sci. Technol. 2017, 51, 7018–7027. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, R. Critical review of low-density polyethylene’s partitioning and diffusion coefficients for trace organic contaminants and implications for its use as a passive sampler. Environ. Sci. Technol. 2015, 49, 3985. [Google Scholar] [CrossRef] [PubMed]
- Apell, J.N.; Gschwend, P.M. Validating the use of performance reference compounds in passive samplers to assess porewater concentrations in sediment beds. Environ. Sci. Technol. 2014, 48, 10301–10307. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.B.; Suresh, P.; Rao, C.; Poyer, I.C.; Nandy, S.; Mallavarapu, M.; Naidu, R.; Davis, G.B.; Patterson, B.M.; Annable, M.D.; et al. Integration of traditional and innovative characterization techniques for flux-based assessment of dense non-aqueous phase liquid (DNAPL) sites. J. Contam. Hydrol. 2009, 105, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Brooks, M.C.; Wood, A.L.; Annable, M.D.; Hatfield, K.; Cho, J.; Holbert, C.; Rao, R.S.C.; Enfield, C.G.; Lynch, K.; Smith, R.E. Changes in contaminant mass discharge from dnapl source mass depletion: Evaluation at two field sites. J. Contam. Hydrol. 2008, 102, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.; Annable, M.; Cho, J.H.; Rao, P.S.C.; Klammler, H. A direct passive method for measuring water and contaminant fluxes in porous media. J. Contam. Hydrol. 2004, 75, 155–181. [Google Scholar] [CrossRef] [PubMed]
- Annable, M.D.; Hatfield, K.; Cho, J.; Klammler, H.; Parker, B.L.; Cherry, J.A.; Rao, P.S.C. Field-scale evaluation of the passive flux meter for simultaneous measurement of groundwater and contaminant fluxes. Environ. Sci. Technol. 2005, 39, 7194–7201. [Google Scholar] [CrossRef] [PubMed]
- Klammler, H.; Hatfield, K.; Annable, M.D. Concepts for measuring horizontal groundwater flow directions using the passive flux meter. Adv. Water Resour. 2007, 30, 984–997. [Google Scholar] [CrossRef]
- Klammler, H.; Hatfield, K.; da Luz, J.A.G.; Annable, M.D.; Newman, M.; Cho, J.; Peacock, A.; Stucker, V.; Ranville, J.; Cabaniss, S.A.; et al. Contaminant discharge and uncertainty estimates from passive flux meter measurements. Water Resour. Res. 2012, 48, 19. [Google Scholar] [CrossRef]
- Padowski, J.C.; Rothfus, E.A.; Jawitz, J.W.; Klammler, H.; Hatfield, K.; Annable, M.D. Effect of passive surface water flux meter design on water and solute mass flux estimates. J. Hydrol. Eng. 2009, 14, 1334–1342. [Google Scholar] [CrossRef]
- Layton, L.; Klammler, H.; Hatfield, K.; Cho, J.; Newman, M.A.; Annable, M.D. Development of a passive sensor for measuring vertical cumulative water and solute mass fluxes in lake sediments and streambeds. Adv. Water Resour. 2017, 105, 1–12. [Google Scholar] [CrossRef]
- Klammler, H.; Hatfield, K.; Newman, M.A.; Cho, J.; Annable, M.D.; Parker, B.L.; Cherry, J.A.; Perminova, I. A new device for characterizing fracture networks and measuring groundwater and contaminant fluxes in fractured rock aquifers. Water Resour. Res. 2016, 52, 5400–5420. [Google Scholar] [CrossRef]
- Kunz, J.V.; Annable, M.D.; Cho, J.; von Tumpling, W.; Hatfield, K.; Rao, S.; Borchardt, D.; Rode, M. Quantifying nutrient fluxes with a new hyporheic passive flux meter (HPFM). Biogeosciences 2017, 14, 631–649. [Google Scholar] [CrossRef]
- Lee, J.; Rao, P.S.C.; Poyer, I.C.; Toole, R.M.; Annable, M.D.; Hatfield, K. Oxyanion flux characterization using passive flux meters: Development and field testing of surfactant-modified granular activated carbon. J. Contam. Hydrol. 2007, 92, 208–229. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.J.; Hatfield, K.; Klammler, H.; Annable, M.D.; Rao, P.S.C. Magnitude and directional measures of water and Cr(VI) fluxes by passive flux meter. Environ. Sci. Technol. 2006, 40, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Verreydt, G.; Annable, M.D.; Kaskassian, S.; Van Keer, I.; Bronders, J.; Diels, L.; Vanderauwera, P. Field demonstration and evaluation of the passive flux meter on a CAH groundwater plume. Environ. Sci. Pollut. Res. 2013, 20, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.D.; Davis, G.B.; Bastow, T.P.; Woodbury, R.J.; Rao, P.S.C.; Annable, M.D.; Rhodes, S. Mass discharge assessment at a brominated DNAPL site: Effects of known DNAPL source mass removal. J. Contam. Hydrol. 2014, 164, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.J.; Hatfield, K.; Annable, M.D.; Gupta, P.; Chirenje, T. Estimation of arsenic contamination in groundwater by the passive flux meter. Environ. Forensics 2005, 6, 77–82. [Google Scholar] [CrossRef]
- Cho, J.Y.; Annable, M.D.; Jawitz, J.W.; Hatfield, K. Passive flux meter measurement of water and nutrient flux in saturated porous media: Bench-scale laboratory tests. J. Environ. Qual. 2007, 36, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.V.; Annable, M.D.; Rao, S.; Rode, M.; Borchardt, D. Hyporheic passive flux meters reveal inverse vertical zonation and high seasonality of nitrogen processing in an anthropogenically modified stream (Holtemme, Germany). Water Resour. Res. 2017, 53, 10155–10172. [Google Scholar] [CrossRef]
- Stucker, V.; Ranville, J.; Newman, M.; Peacock, A.; Cho, J.; Hatfield, K. Evaluation and application of anion exchange resins to measure groundwater uranium flux at a former uranium mill site. Water Res. 2011, 45, 4866–4876. [Google Scholar] [CrossRef] [PubMed]
- Zenker, M.J.; Borden, R.C.; Barlaz, M.A. Occurrence and treatment of 1,4-dioxane in aqueous environments. Environ. Eng. Sci. 2003, 20, 423–432. [Google Scholar] [CrossRef]
- Zhang, S.; Gedalanga, P.B.; Mahendra, S. Advances in bioremediation of 1,4-dioxane-contaminated waters. J. Environ. Manag. 2017, 204, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.J.J.; Illman, W.A. Bioremediation and Natural Attenuation; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Howard, P. Handbook of Environmental Fate and Exposure Data for Organic Chemicals; Lewis Publishers, Inc.: Chelsea, MI, USA, 1990. [Google Scholar]
- ITRC. Protocol for Use of Five Passive Sampler to Sample for a Variety of Contaminants in Groundwater; ITRC: Washington, DC, USA, 2007.
- Paquet, L.; Monteil-Rivera, F.; Hatzinger, P.B.; Fuller, M.E.; Hawari, J. Analysis of the key intermediates of RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine) in groundwater: Occurrence, stability and preservation. J. Environ. Monit. 2011, 13, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.C.; Myers, K.F.; Brannon, J.M.; Delfino, J.J. Effects of pH and temperature on the aqueous solubility and dissolution rate of 2,4,6-trinitrotoluene (TNT), hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX), and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX). J. Chem. Eng. Data 2001, 46, 1549–1555. [Google Scholar] [CrossRef]
- Pascoe, G.A.; Kroeger, K.; Leisle, D.; Feldpausch, R.J. Munition constituents: Preliminary sediment screening criteria for the protection of marine benthic invertebrates. Chemosphere 2010, 81, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.H.; Smith, P.N.; Anderson, T.A. Evaluating the bioavailability of explosive metabolites, hexahydro-1-nitroso-3,5-dinitro-1,3,5-triazine (MNX) and hexahydro-1,3,5-trinitroso-1,3,5-triazine (TNX), in soils using passive sampling devices. J. Chromatogr. A 2006, 1101, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.; Wild, B.; George, R.D.; Belden, J.B.; Lotufo, G.R. Optimization and field demonstration of a passive sampling technology for monitoring conventional munition constituents in aquatic environments. Mar. Technol. Soc. J. 2016, 50, 23–32. [Google Scholar] [CrossRef]
- Morley, M.C.; Henke, J.L.; Speitel, G.E. Adsorption of rdx and hmx in rapid small-scale column tests: Implications for full-scale adsorbers. J. Environ. Eng.-ASCE 2005, 131, 29–37. [Google Scholar] [CrossRef]
- Heilmann, H.M.; Wiesmann, U.; Stenstrom, M.K. Kinetics of the alkaline hydrolysis of high explosives RDX and HMX in aqueous solution and adsorbed to activated carbon. Environ. Sci. Technol. 1996, 30, 1485–1492. [Google Scholar] [CrossRef]
- Millerick, K.; Drew, S.R.; Finneran, K.T. Electron shuttle-mediated biotransformation of hexahydro-1,3,5-trinitro-1,3,5-triazine adsorbed to granular activated carbon. Environ. Sci. Technol. 2013, 47, 8743–8750. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Mueller, J.G.; Seech, A.G.; Henderson, J.K.; Wilson, J.T. Interactions between biological and abiotic pathways in the reduction of chlorinated solvents. Remedation 2009, 20, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Butler, E.C.; Hayes, K.F. Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide. Environ. Sci. Technol. 1999, 33, 2021–2027. [Google Scholar] [CrossRef]
- Butler, E.C.; Hayes, K.F. Factors influencing rates and products in the transformation of trichloroethylene by iron sulfide and iron metal. Environ. Sci. Technol. 2001, 35, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Ferrey, M.L.; Wilkin, R.T.; Ford, R.G.; Wilson, J.T. Nonbiological removal of cis-dichloroethylene and 1,1-dichloroethylene in aquifer sediment containing magnetite. Environ. Sci. Technol. 2004, 38, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Hayes, K.F. Reductive dechlorination of tetrachloroethylene and trichloroethylene by mackinawite (FeS) in the presence of metals: Reaction rates. Environ. Sci. Technol. 2007, 41, 6390–6396. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Anantharaman, K.; Han, Y.S.; Hayes, K.F. Abiotic reductive dechlorination of cis-dichloroethylene by Fe species formed during iron- or sulfate-reduction. Environ. Sci. Technol. 2011, 45, 5186–5194. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Batchelor, B. Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals. 1. Pyrite and magnetite. Environ. Sci. Technol. 2002, 36, 5147–5154. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Batchelor, B. Abiotic, reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals. 2. Green rust. Environ. Sci. Technol. 2002, 36, 5348–5354. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.J.; Nguyen, D.; Chappell, R.W.; Whiting, K.; Gillette, J.; Bodour, A.; Wilson, J.T. Factors controlling in situ biogeochemical transformation of trichloroethene: Column study. Ground Water Monit. Remed. 2014, 34, 65–78. [Google Scholar] [CrossRef]
- Whiting, K.; Evans, P.J.; Lebron, C.; Henry, B.; Wilson, J.T.; Becvar, E. Factors controlling in situ biogeochemical transformation of trichloroethene: Field survey. Ground Water Monit. Remed. 2014, 34, 79–94. [Google Scholar] [CrossRef]
- Kennedy, L.; Everett, J.W.; Gonzales, J. Aqueous and mineral intrinsic bioremediation assessment: Natural attenuation. J. Environ. Eng.-ASCE 2004, 130, 942–950. [Google Scholar] [CrossRef]
- Wilkin, R.T.; Bischoff, K.J. Coulometric determination of total sulfur and reduced inorganic sulfur fractions in environmental samples. Talanta 2006, 70, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeier, T.H.; Swanson, M.A.; Moutoux, D.E.; Gordon, E.K.; Wilson, J.T.; Wilson, B.H.; Kampbell, D.H.; Haas, P.E.; Miller, R.N.; Hansen, J.E.; et al. Technical Protocol for Evaluating Natural Attenuation of Chlorinated Solvents in Ground Water; United States Environmental Protection Agency: Washington, DC, USA, 1998.
- Nobre, R.C.M.; Nobre, M.M.M.; Campos, T.M.P.; Ogles, D. In-situ biodegradation potential of 1,2-DCA and VC at sites with different hydrogeological settings. J. Hazard. Mater. 2017, 340, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.L. Epistemology of environmental microbiology. Environ. Sci. Technol. 1998, 32, 429–439. [Google Scholar] [CrossRef]
- Baruthio, F. Toxic effects of chromium and its compounds. Biol. Trace Elem. Res. 1992, 32, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Grevatt, P.C. Toxicological Review of Trivalent Chromium; Agency, USEP: Washington, DC, USA, 1998.
- Driscoll, S.K.; McArdle, M.E.; Plumlee, M.H.; Proctor, D. Evaluation of hexavalent chromium in sediment pore water of the Hackensack river, New Jersey, USA. Environ. Toxicol. Chem. 2010, 29, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Hopp, L.; Peiffer, S.; Durner, W. Spatial variability of arsenic and chromium in the soil water at a former wood preserving site. J. Contam. Hydrol. 2006, 85, 159–178. [Google Scholar] [CrossRef] [PubMed]
- James, B.R. The challenge of remediating chromium-contaminated soil. Environ. Sci. Technol. 1996, 30, A248–A251. [Google Scholar] [CrossRef] [PubMed]
- Barbee, G.C.; Brown, K.W. Comparison between suction and free-drainage soil solution samplers. Soil Sci. 1986, 141, 149–154. [Google Scholar] [CrossRef]
- Basu, N.B.; Rao, P.S.C.; Poyer, I.C.; Annable, M.D.; Hatfield, K. Flux-based assessment at a manufacturing site contaminated with trichloroethylene. J. Contam. Hydrol. 2006, 86, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Johns, M.M.; Marshall, W.E.; Toles, C.A. Agricultural by-products as granular activated carbons for adsorbing dissolved metals and organics. J. Chem. Technol. Biotechnol. 1998, 71, 131–140. [Google Scholar] [CrossRef]
- Otto, M.; Nagaraja, S. Treatment technologies for 1,4-dioxane: Fundamentals and field applications. Remedation 2007, 17, 81–88. [Google Scholar] [CrossRef]
- Navalon, S.; Alvaro, M.; Garcia, H. Analysis of organic compounds in an urban wastewater treatment plant effluent. Environ. Technol. 2011, 32, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Zabkova, M.; Rodrigues, A.E. Comparative study of the adsorption of phenol and salicylic acid from aqueous solution onto nonionic polymeric resins. Sep. Purif. Technol. 2005, 45, 86–95. [Google Scholar] [CrossRef]
- Maloney, S.W.; Adrian, N.R.; Hickey, R.F.; Heine, R.L. Anaerobic treatment of pinkwater in a fluidized bed reactor containing GAC. J. Hazard. Mater. 2002, 92, 77–88. [Google Scholar] [CrossRef]
- Parette, R.; Cannon, F.S.; Weeks, K. Removing low ppb level perchlorate, RDX, and HMX from groundwater with cetyltrimethylammonium chloride (CTAC) pre-loaded activated carbon. Water Res. 2005, 39, 4683–4692. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Dixon, J.B. Oxidation and fate of chromium in soils. Soil Sci. Plant Nutr. 2002, 48, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Daoud, W.; Ebadi, T.; Fahimifar, A. Optimization of hexavalent chromium removal from aqueous solution using acid-modified granular activated carbon as adsorbent through response surface methodology. Korean J. Chem. Eng. 2015, 32, 1119–1128. [Google Scholar] [CrossRef]
- Di Natale, F.; Lancia, A.; Molino, A.; Musmarra, D. Removal of chromium ions form aqueous solutions by adsorption on activated carbon and char. J. Hazard. Mater. 2007, 145, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Fetter, C.W. Contaminant Hydrology; Prentice Hall: Upper Saddle River, NJ, USA, 1999. [Google Scholar]
- Han, I.; Schlautman, M.A.; Batchelor, B. Removal of hexavalent chromium from groundwater by granular activated carbon. Water Environ. Res. 2000, 72, 29–39. [Google Scholar] [CrossRef]
- Piazzoli, A.; Antonelli, M. Feasibility assessment of chromium removal from groundwater for drinking purposes by sorption on granular activated carbon and strong base anion exchange. Water Air Soil Pollut. 2018, 229, 17. [Google Scholar] [CrossRef]
- Satapathy, D.; Natarajan, G.S.; Patil, S.J. Adsorption characteristics of chromium(VI) on granular activated carbon. J. Chin. Chem. Soc. 2005, 52, 35–44. [Google Scholar] [CrossRef]
- Singha, S.; Sarkar, U. Analysis of the dynamics of a packed column using semi-empirical models: Case studies with the removal of hexavalent chromium from effluent wastewater. Korean J. Chem. Eng. 2015, 32, 20–29. [Google Scholar] [CrossRef]
- Song, H.O.; Yao, Z.J.; Shuang, C.D.; Li, A.M. Accelerated removal of nitrate from aqueous solution by utilizing polyacrylic anion exchange resin with magnetic separation performance. J. Ind. Eng. Chem. 2014, 20, 2888–2894. [Google Scholar] [CrossRef]
- Song, H.O.; Yao, Z.J.; Wang, M.Q.; Wang, J.N.; Zhu, Z.L.; Li, A.M. Effect of dissolved organic matter on nitrate-nitrogen removal by anion exchange resin and kinetics studies. J. Environ. Sci. 2013, 25, 105–113. [Google Scholar] [CrossRef]
- Primo, O.; Rivero, M.J.; Urtiaga, A.M.; Ortiz, I. Nitrate removal from electro-oxidized landfill leachate by ion exchange. J. Hazard. Mater. 2009, 164, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, D.; Leao, V.A. Fundamental aspects related to batch and fixed-bed sulfate sorption by the macroporous type 1 strong base ion exchange resin Purolite A500. J. Environ. Manag. 2014, 145, 106–112. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breakthrough | Methanol | 1,4-Dioxane | Methylene Chloride | cis-1,2-Dichloroethene | ||||
|---|---|---|---|---|---|---|---|---|
| Mass Retained by GAC | R Factor | Mass Retained by GAC | R Factor | Mass Retained by GAC | R Factor | Mass Retained by GAC | R Factor | |
| Initial (mg/g) | 0.04 | 1 | 6.4 | 62 | 16.6 | 147 | 71.3 | 713 |
| 50% (mg/g) | 0.12 | 3 | 13.3 | 192 | 28.2 | 455 | 98.1 | 1700 |
| 100% (mg/g) | 0.4 | 5 | 19.8 | 235 | 43.6 | 386 | 122.5 | 1167 |
| Well | PFM Flux-Averaged Concentration (μg/L) | Measured Aqueous Phase Concentration (μg/L) | Percent Difference (%) |
|---|---|---|---|
| PFM 1 | <5 | 2.3 | - |
| PFM 2 | <5 | 330 | - |
| PFM 3 | 18 | 8.6 | 68 |
| PFM 4 | <5 | 1.9 | - |
| PFM 5 | 5932 | 1600 | 115 |
| PFM 6 | 1663 | 1700 | 2 |
| PFM 7 | 2195 | 990 | 76 |
| PFM 8 | 1675 | 480 | 111 |
| PFM 9 | 55 | 46 | 18 |
| PFM 10 | 20 | <1 | - |
| PFM 11 | <5 | 2.3 | - |
| PFM 12 | 1384 | 280 | 133 |
| PFM 13 | 602 | 310 | 64 |
| Breakthrough | Sulfate | |
|---|---|---|
| Breakthrough (mg/g) | Retardation Factor | |
| Initial | 50.5 | 27 |
| 50% | 68 | 15 |
| 100% | 70.4 | 37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haluska, A.A.; Thiemann, M.S.; Evans, P.J.; Cho, J.; Annable, M.D. Expanded Application of the Passive Flux Meter: In-Situ Measurements of 1,4-Dioxane, Sulfate, Cr(VI) and RDX. Water 2018, 10, 1335. https://doi.org/10.3390/w10101335
Haluska AA, Thiemann MS, Evans PJ, Cho J, Annable MD. Expanded Application of the Passive Flux Meter: In-Situ Measurements of 1,4-Dioxane, Sulfate, Cr(VI) and RDX. Water. 2018; 10(10):1335. https://doi.org/10.3390/w10101335
Chicago/Turabian StyleHaluska, Alexander A., Meghan S. Thiemann, Patrick J. Evans, Jaehyun Cho, and Michael D. Annable. 2018. "Expanded Application of the Passive Flux Meter: In-Situ Measurements of 1,4-Dioxane, Sulfate, Cr(VI) and RDX" Water 10, no. 10: 1335. https://doi.org/10.3390/w10101335
APA StyleHaluska, A. A., Thiemann, M. S., Evans, P. J., Cho, J., & Annable, M. D. (2018). Expanded Application of the Passive Flux Meter: In-Situ Measurements of 1,4-Dioxane, Sulfate, Cr(VI) and RDX. Water, 10(10), 1335. https://doi.org/10.3390/w10101335