NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B

Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Catalyst
2.1.1. Preparation of graphene oxide(GO)
2.1.2. Preparation of Ni-Fe-LDH-rGO
2.1.3. Preparation of NiO-NiFe2O4-rGO
2.2. Characterization of Catalyst
2.3. Catalytic Test Procedures and Analytical Method
3. Results and Discussion
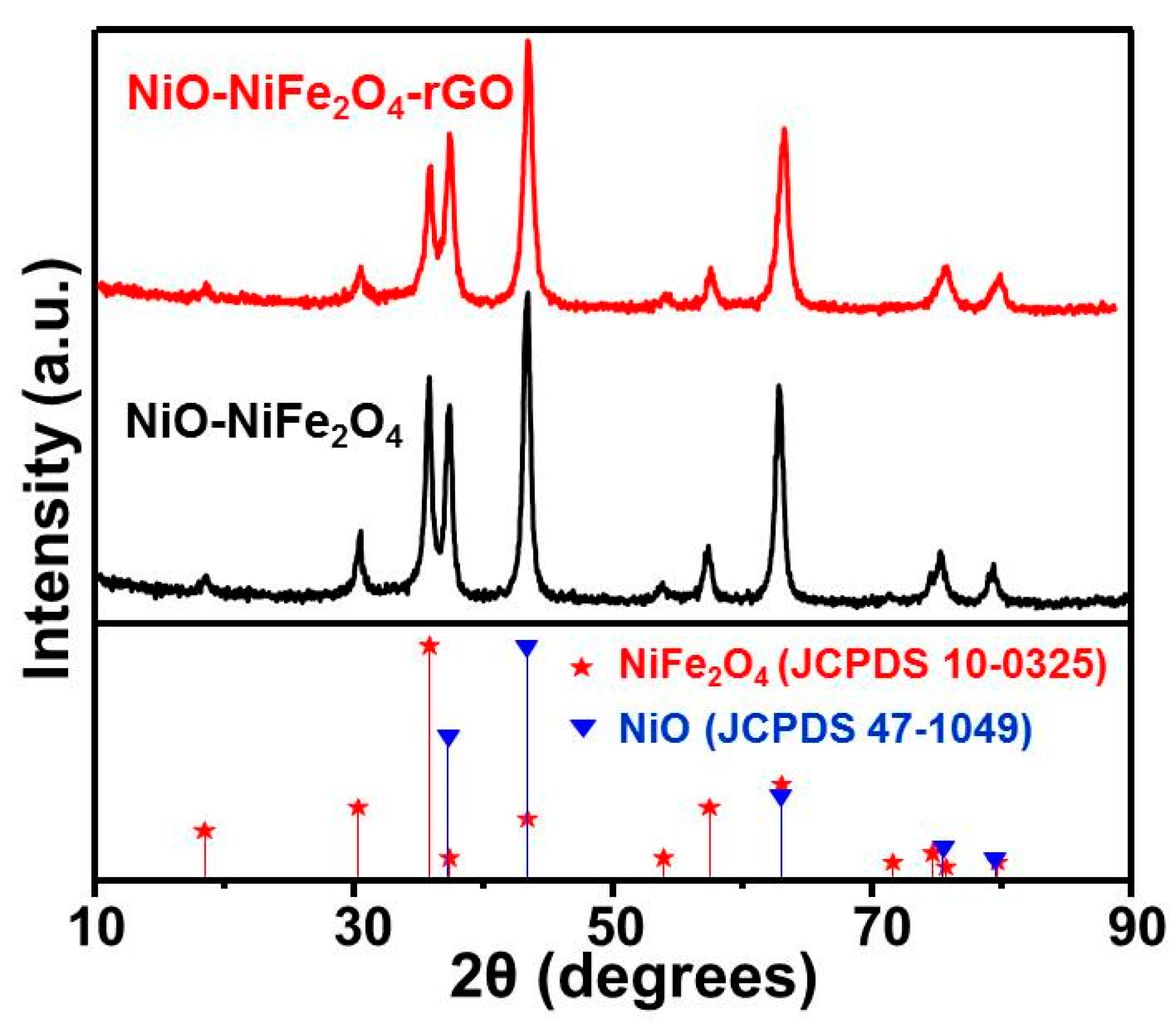
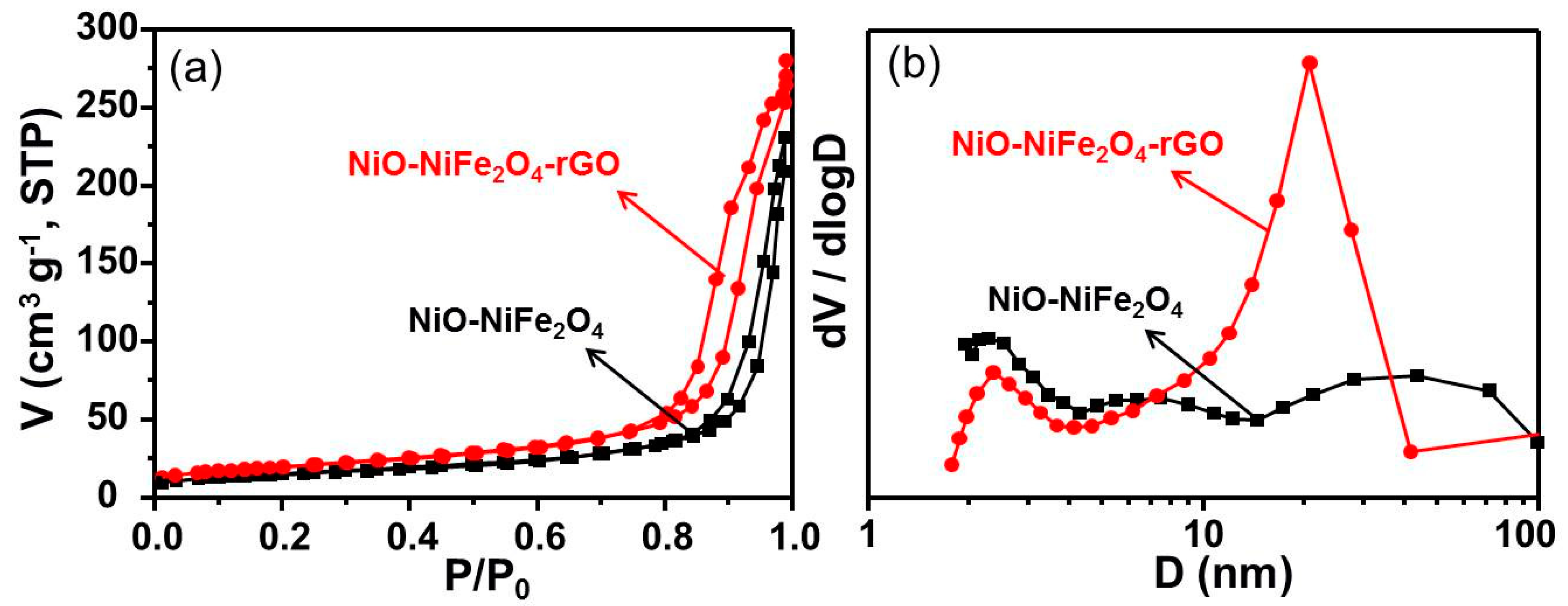
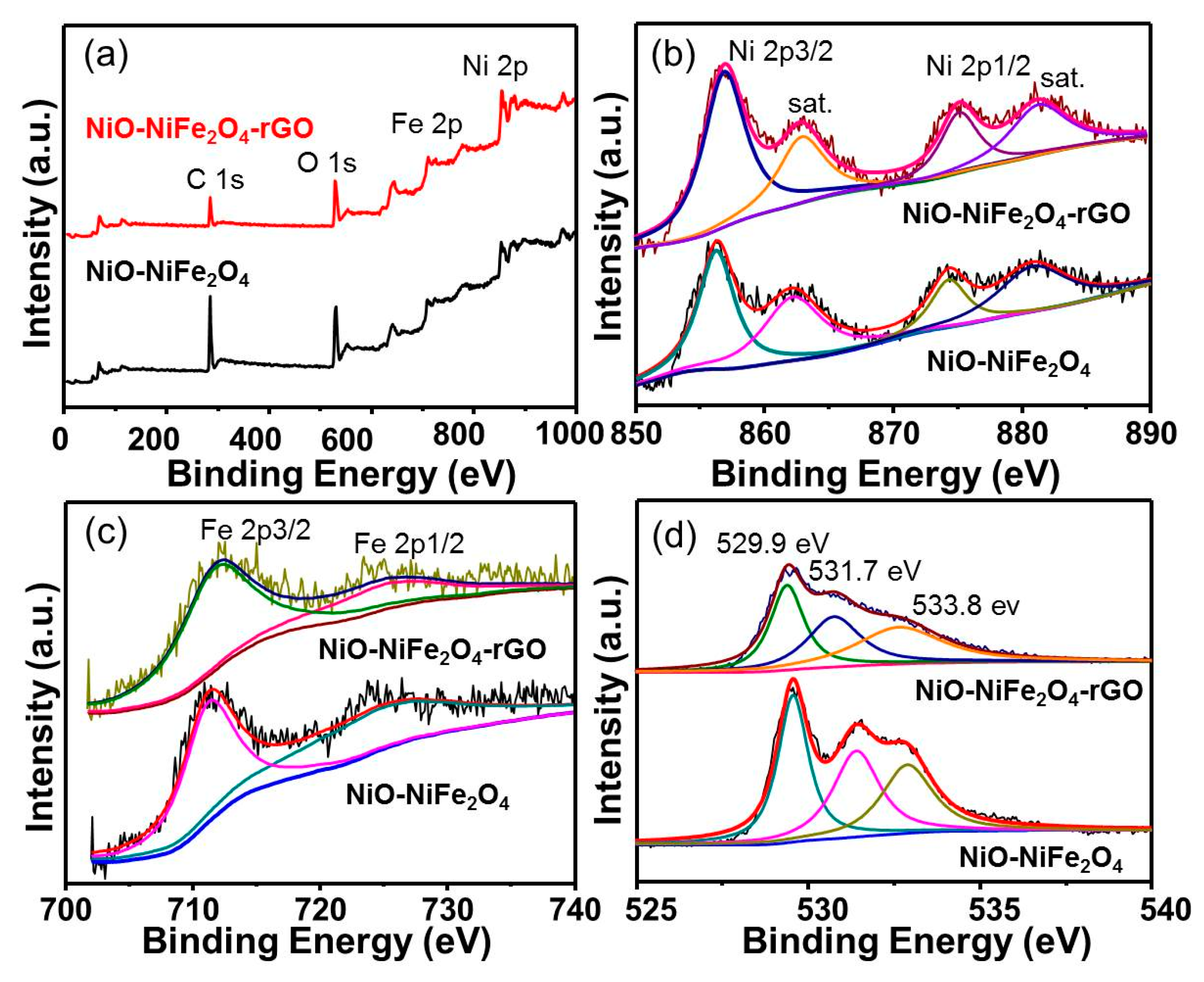
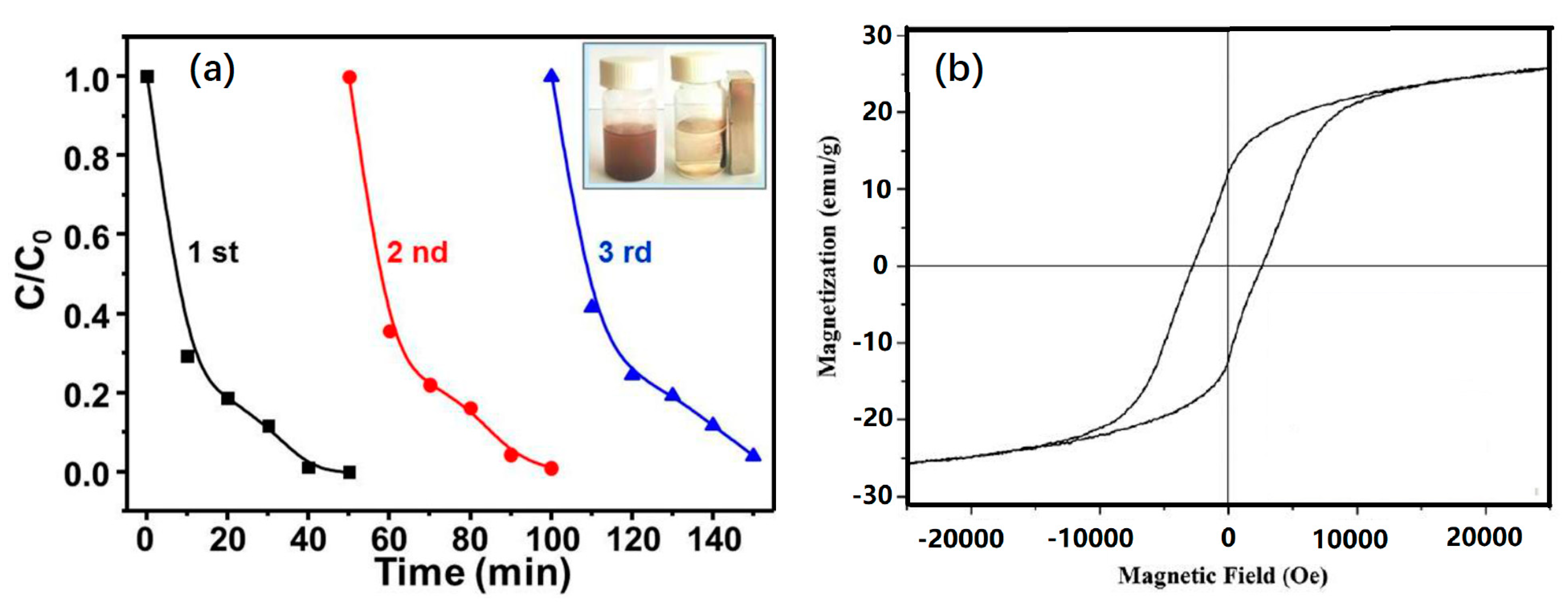
3.1. Catalyst Characterization
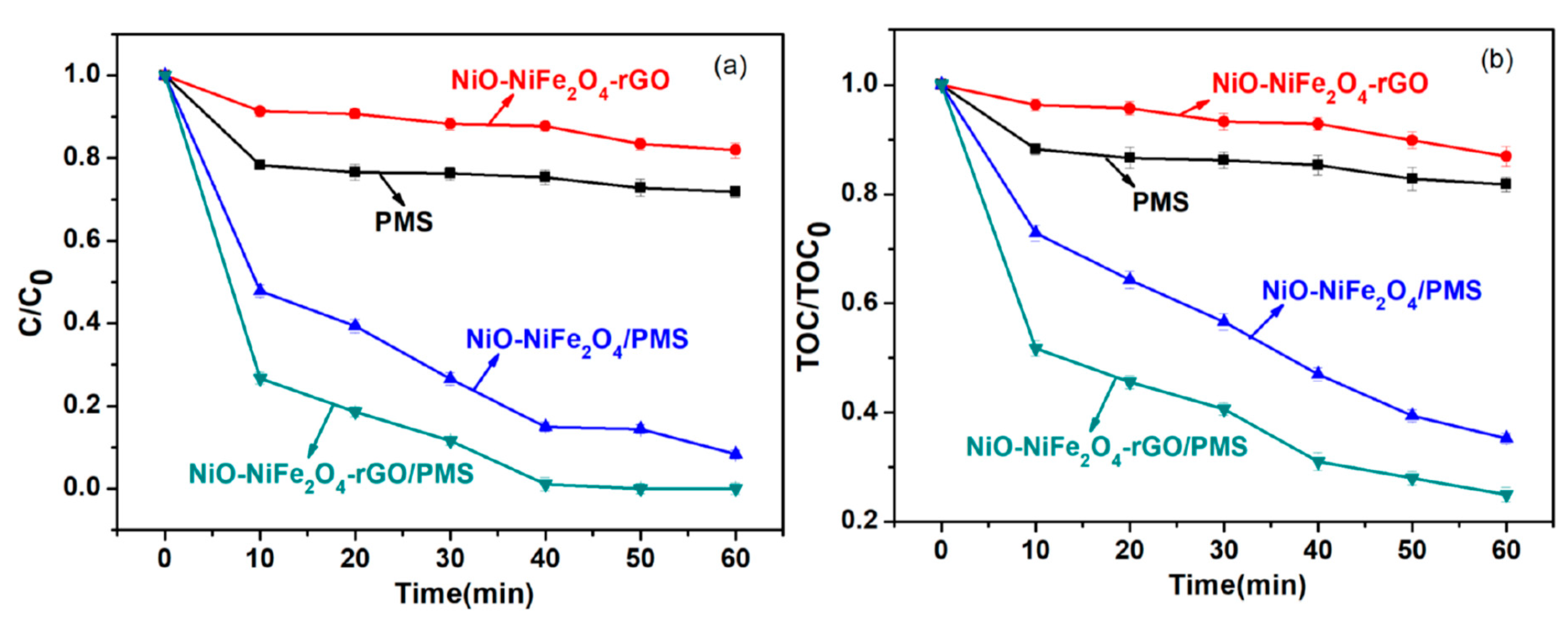
3.2. Activation of PMS by NiO-NiFe2O4-rGO for RhB Degradation
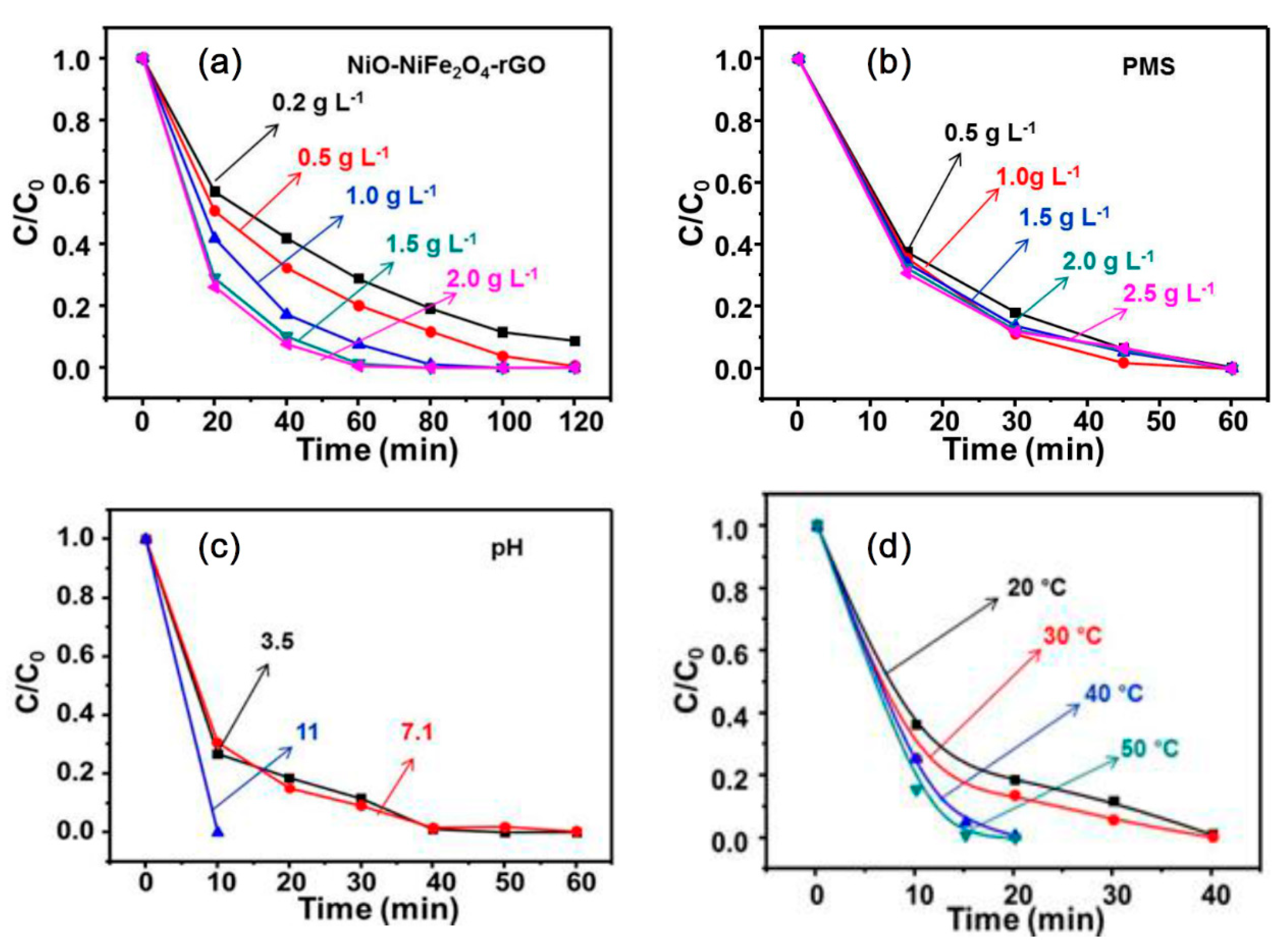
3.3. The Effect of Different Process Parameters on the RhB Degradation
3.3.1. The Effect of NiO-NiFe2O4-rGO Dosage on RhB Degradation
3.3.2. Effect of PMS Dosage on RhB Degradation
3.3.3. Effect of Initial pH on RhB Degradation
3.3.4. The Effect of the Temperature on RhB Degradation
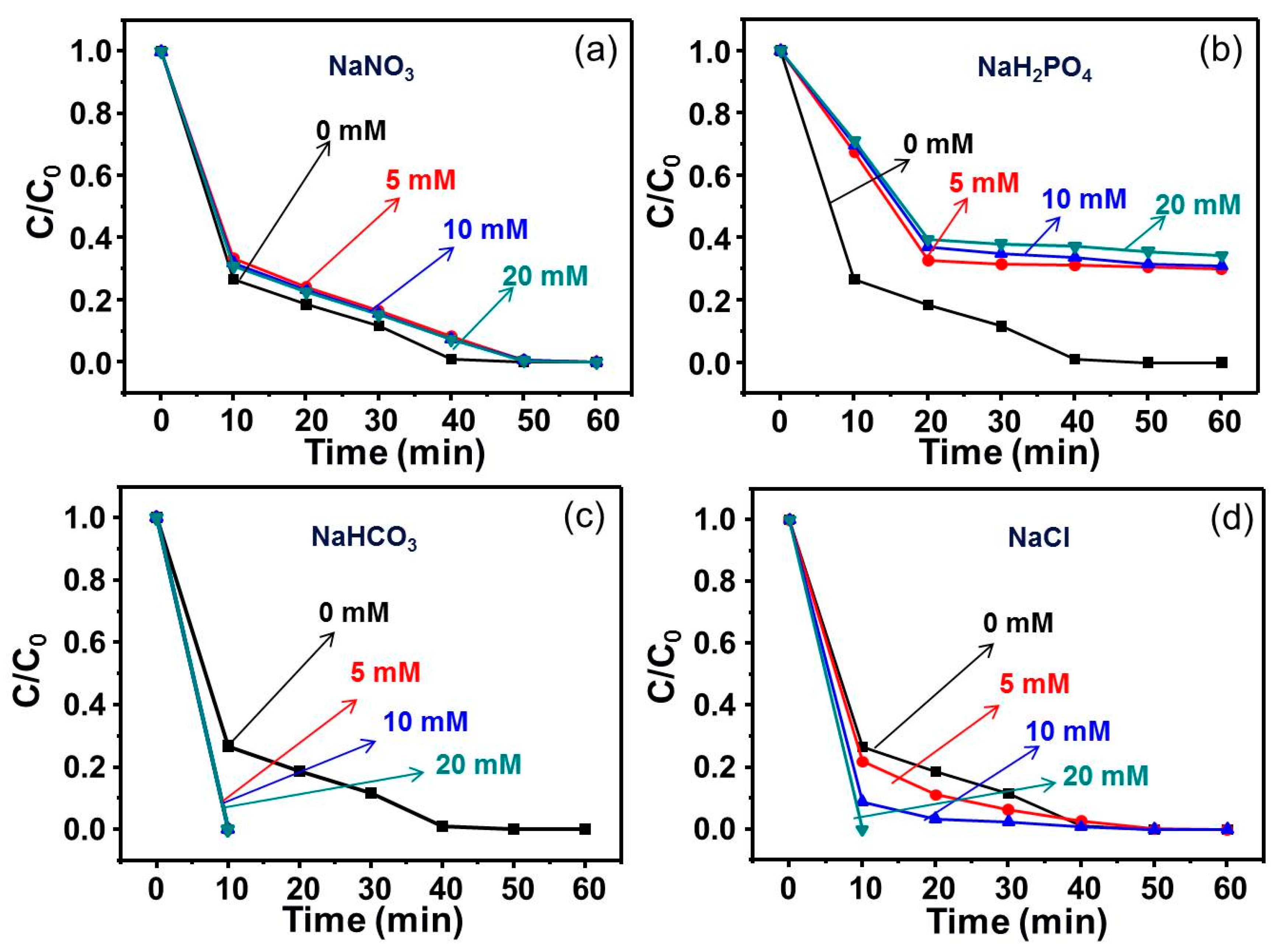
3.4. Effect of Common Anions on RhB Degradation
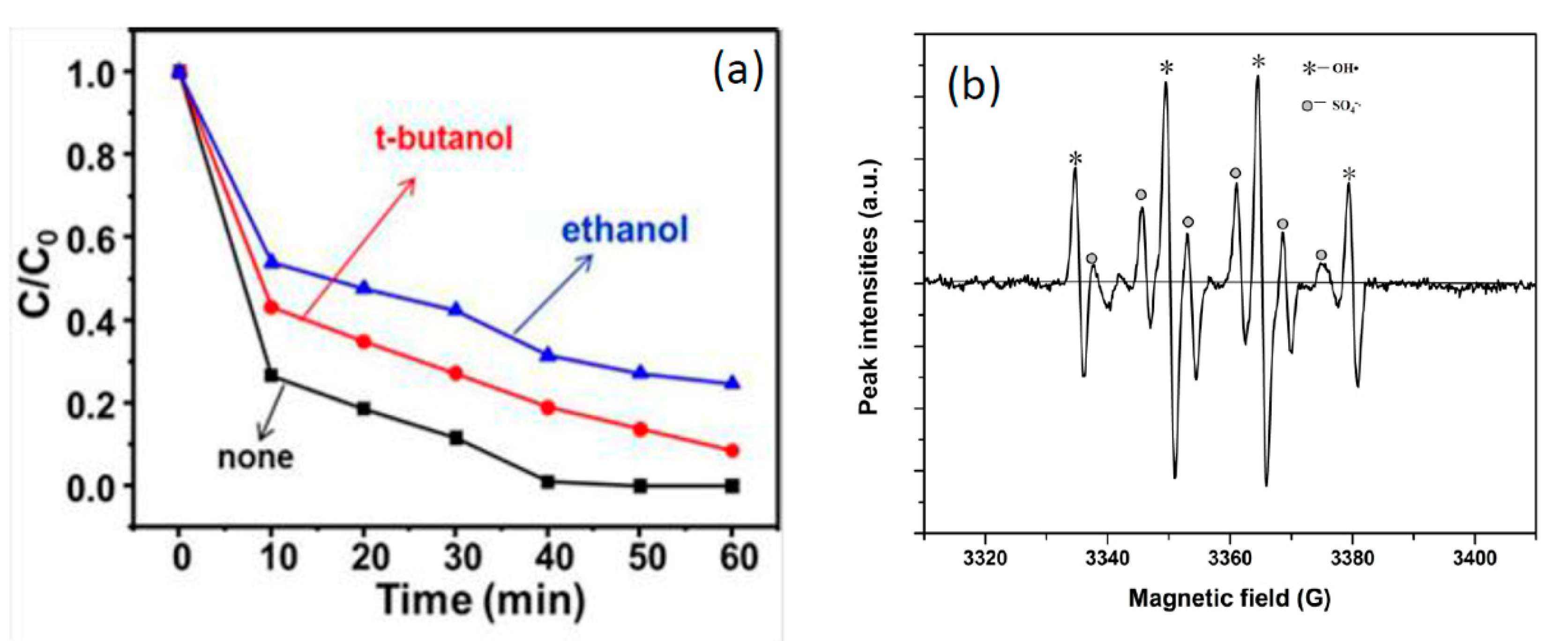
3.5. Mechanism of PMS Activation in NiO-NiFe2O4-rGO/PMS System
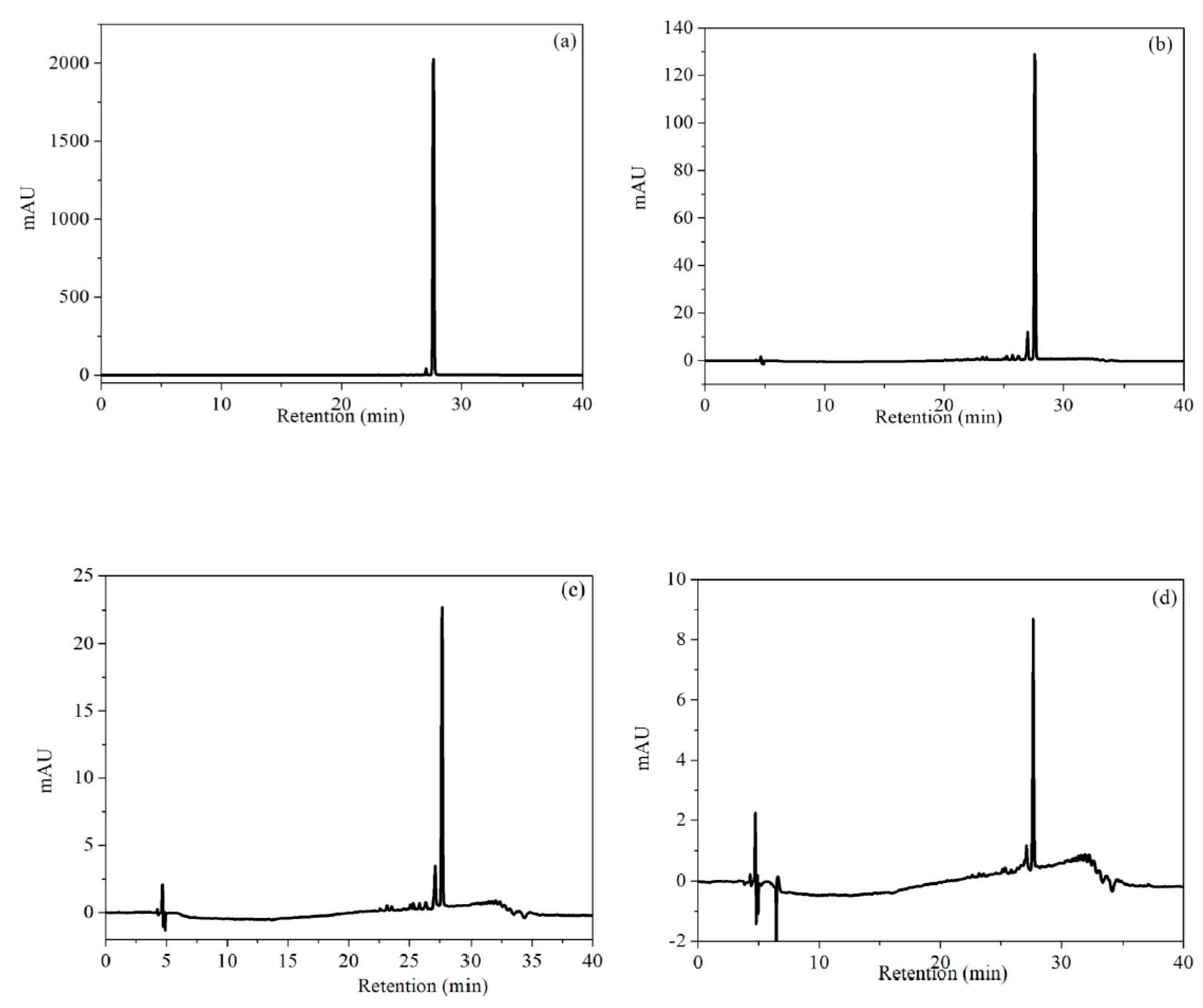
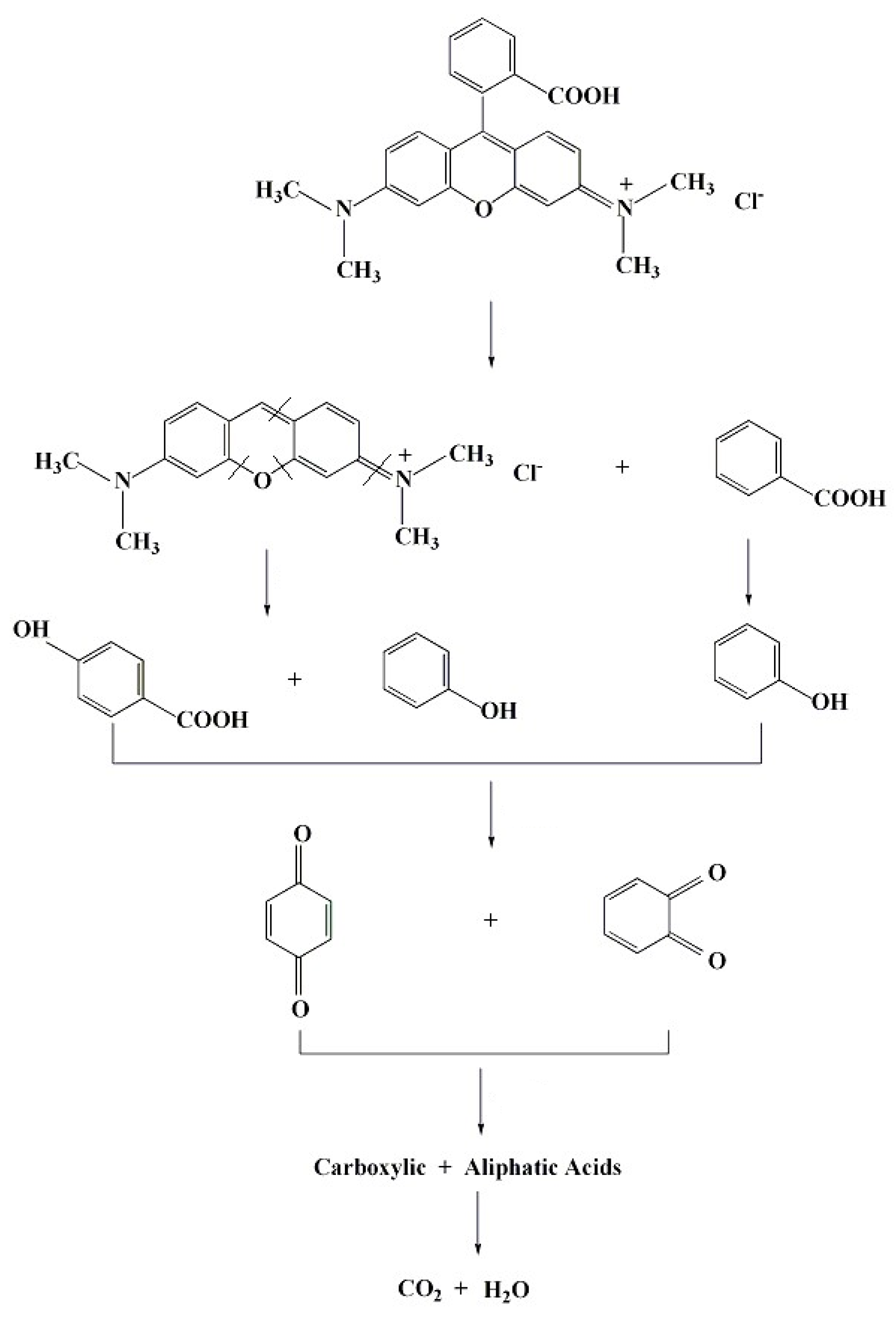
3.6. Degradation Pathway of RhB in NiO-NiFe2O4-rGO/PMS System
3.7. Recyclability of NiO-NiFe2O4-rGO to Activate PMS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bili´nskaa, L.; Gmurek, M.; Ledakowicz, S. Textile wastewater treatment by AOPs for brine reuse. Process Saf. Environ. 2017, 109, 420–428. [Google Scholar] [CrossRef]
- Rosario-Ortiz, F.L.; Wert, E.C.; Snyder, S.A. Evaluation of UV/H2O2 treatment for the oxidation of pharmaceuticals in wastewater. Water Res. 2010, 44, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.-A.; Hofmann, R. A comparative study of the hydroxyl radical scavenging capacity of activated sludge and membrane bioreactor wastewater effluents. Water Sci. Technol. 2016, 73, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Cheng, M.; Zeng, G.; Huang, D.; Lai, C.; Lui, Y.; Zhang, C.; Wan, J.; Hu, L.; Zhou, C.; Xiong, W. Efficient degradation of sulfamethazine in simulated and real wastewaterat slightly basic pH values using Co-SAM-SCS/H2O2 fenton-like system. Water Res. 2018, 138, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Mahdi-Ahmed, M.; Chiron, S. Ciprofloxacin oxidation by UV-C activated peroxymonosulfate in wastewater. J. Hazard. Mater. 2014, 265, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Babuponnusami, A.; Muthukumar, K. A review on Fenton and improvements to the Fenton process for wastewater treatment. J. Environ. Chem. Eng. 2014, 2, 557–572. [Google Scholar] [CrossRef]
- Zhang, B.T.; Zhang, Y.; Teng, Y.H.; Fan, M.H. Sulfate radical and its application in decontamination technologies. Crit. Rev. Env. Sci. Tec. 2015, 45, 1756–1800. [Google Scholar] [CrossRef]
- Fan, G.; Li, F.; Evans, D.G. Catalytic applications of layered double hydroxides: Recent advances and perspectives. Chem. Soc. Rev. 2014, 43, 7040–7066. [Google Scholar] [CrossRef] [PubMed]
- Monaam, B.; Alexandre, B.; Ahmed, A.; Brigitte, S.; Habib, E.; Mokhtar, F.; Sabine, S.; Rabah, B. Co2SnO4 nanoparticles as a high performance catalyst for oxidative degradation of rhodamine B dye and pentachlorophenol by activation of peroxymonosulfate. Phys. Chem. Chem. Phys. 2017, 19, 6569–6578. [Google Scholar]
- Ren, Y.; Lin, L.; Ma, J.; Yang, J.; Feng, J.; Fan, Z. Sulfate radicals induced from peroxymonosulfate by magnetic ferro spinel MFe2O4(M = Co, Cu Mn, and Zn) as heterogeneous catalysts in the water. Appl. Catal. B 2015, 165, 572–578. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Li, M.; Fan, J.; Zhao, G. Magnetic ordered mesoporous copper ferrite as a heterogeneous Fenton catalyst for the degradation of imidacloprid. Appl. Catal. B 2014, 147, 534–545. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, Y.; Lu, F.; Wei, F.; Wang, X.; Wang, S. Magnetic recoverable MnFe2O4 and MnFe2O4-graphene hybrid as heterogeneous catalysts of peroxymonosulfate activation for efficient degradation of aqueous organic pollutants. J. Hazard. Mater. 2014, 270, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Chu, W.; Gan, L. Environmental application of graphene-based CoFe2O4 as an activator of peroxymonosulfate for the degradation of a plasticizer. Chem. Eng. J. 2015, 263, 435–443. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Zong, M.; Huang, Y.; Ding, X. One-step hydrothermal synthesis and microwave electromagnetic properties of RGO/NiFe2O4 composite. Ceram. Int. 2014, 40, 6821–6828. [Google Scholar] [CrossRef]
- Fu, M.; Jiao, Q.; Zhao, Y. Preparation of NiFe2O4 nanorod-graphene composites via an ionic liquid assisted one-step hydrothermal approach and their microwave absorbing properties. J. Mater. Chem. A 2013, 1, 5577–5586. [Google Scholar] [CrossRef]
- Liao, Y.; Fu, M.; Chen, L.; Wu, J.; Huang, B.; Ye, D. Catalytic oxidation of toluene over nanorod-structured Mn–Ce mixed oxides. Catal. Today 2013, 216, 220–228. [Google Scholar] [CrossRef]
- Ball, D.L.; Edwards, J.O. The kinetics and mechanism of the decomposition of Caro’s acid. J. Am. Chem. Soc. 1956, 78, 1125–1129. [Google Scholar] [CrossRef]
- Rastogi, A.; Al-Abed, S.R.; Dionysiou, D.D. Sulfate radical-based ferrous–peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems. Appl. Catal. B 2009, 85, 171–179. [Google Scholar] [CrossRef]
- Hussain, I.; Zhang, Y.Q.; Huang, S.B. Degradation of aniline with zero-valent iron as an activator of persulfate in aqueous solution. Rsc Adv. 2014, 4, 3502–3511. [Google Scholar] [CrossRef]
- Yang, S.; Wang, P.; Yang, X.; Shan, L.; Zhang, W.; Shao, X.; Niu, R. Degradation efficiencies of azo dye Acid Orange 7 by the interaction of heat, UV and anions with common oxidants: Persulfate, peroxymonosulfate and hydrogen peroxide. J. Hazard. Mater. 2010, 179, 552–558. [Google Scholar] [CrossRef]
- Qi, C.D.; Liu, X.T.; Zhao, W.; Lin, C.Y.; Ma, J.; Shi, W.X.; Sun, Q.; Xiao, H. Degradation and dechlorination of pentachlorophenol by microwave-activated persulfate. Environ. Sci. Pollut. R. 2015, 22, 4670–4679. [Google Scholar] [CrossRef]
- Qi, F.; Chu, W.; Xu, B. Modeling the heterogeneous peroxymonosulfate/Co-MCM41 process for the degradation of caffeine and the study of influence of cobalt sources. Chem. Eng. J. 2014, 235, 10–18. [Google Scholar] [CrossRef]
- Ji, Y.; Dong, C.; Kong, D.; Lu, J. New insights into atrazine degradation by cobalt catalyzed peroxymonosulfate oxidation: Kinetics, reaction products and transformation mechanisms. J. Hazard. Mater. 2015, 285, 491–500. [Google Scholar] [CrossRef]
- Chan, K.H.; Chu, W. Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate: Different cobalt counteranions in homogenous process and cobalt oxide catalysts peroxymonosulfate. Water Res. 2009, 43, 2513–2521. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Ang, H.M.; Tade, M.O.; Wang, S. Facile Synthesis of hierarchically structured magnetic MnO2/ZnFe2O4 hybrid materials and their performance in heterogeneous activation of peroxymonosulfate. ACS Appl. Mater. Interfaces 2014, 6, 19914–19923. [Google Scholar] [CrossRef]
- Wu, T.X.; Liu, G.M.; Zhao, J.C. Photoassisted degradation of dye pollutants. V. Self-photosensitized oxidative transformation of Rhodamine B under visible light irradiation in aqueous TiO2 dispersions. J. Phys. Chem. 1998, 102, 5845–5851. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2·g−1) | Vtotal (cm3·g−1) | Vmic (cm3·g−1) |
|---|---|---|---|
| NiO-NiFe2O4 | 56.29 | 0.36 | 0.005 |
| NiO-NiFe2O4-rGO | 70.6 | 0.43 | 0.004 |
| Temperature (K) | ||||
| 293 | 303 | 313 | 323 | |
| K (min−1) | 0.3012 | 0.2231 | 0.1423 | 0.1003 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Li, Y.; Zhang, G.; Yang, F.; He, P. NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B. Water 2019, 11, 384. https://doi.org/10.3390/w11020384
Xu X, Li Y, Zhang G, Yang F, He P. NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B. Water. 2019; 11(2):384. https://doi.org/10.3390/w11020384
Chicago/Turabian StyleXu, Xiaochen, Yanfang Li, Guoquan Zhang, Fenglin Yang, and Ping He. 2019. "NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B" Water 11, no. 2: 384. https://doi.org/10.3390/w11020384
APA StyleXu, X., Li, Y., Zhang, G., Yang, F., & He, P. (2019). NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B. Water, 11(2), 384. https://doi.org/10.3390/w11020384