1. Introduction
Membrane bioreactor (MBR) and biofilm membrane bioreactor (BF-MBR) are advanced solutions for the problem of water scarcity, which have been recognized as highly competitive technologies when applied in water reuse schemes [
1,
2,
3,
4]. However, membrane fouling remains the major barrier for any MBR and BF-MBR expansion [
5,
6].
With regard to membrane fouling mechanisms, cake and gel layer formation and membrane pore blockage were identified as major contributors to any filtration resistance [
7]. The formation of the cake layer is mainly attributed to the deposition of suspended solids, whose size is bigger than the membrane pores, onto the membrane surface or onto the sealed pores, with a subsequent stacking [
7,
8]. The gel layer is the matrix, which consists of highly concentrated solutes and macromolecular species, deposited at the membrane surface.
This gel layer is usually intertwined with the cake matrix, therefore it is highly complicated to distinguish either of them [
9]. The formation of the cake layer, as well as the gel matrix at the membrane surface, are governed by the pressure-driven convective flow from the bulk mixed liquor solution to the membrane during filtration [
7,
9,
10].
Concentration polarization (CP) is the other type of solute fouling, which accompanies every filtration system, being, however, of marginal importance in the MBR/BF-MBR operation [
9,
11]. This phenomenon is entailed by the tendency of the solutes, rejected by the membrane, to accumulate at the membrane-solution interface within the concentration boundary region, driven by the concentration gradients, and to form a highly concentrated zone, called a concentration polarization layer. In contrast to the cake and gel layers, the transport within the concentration polarization region is diffusion, described by the Fick’s first law [
7,
9,
10]. The solids can diffuse back to the bulk mixed liquor in the CP layer, if they are not entrapped in the gel or cake layer.
The concentration polarization model and the mechanisms and mathematical description of cake layer compaction are explained in detail by Yoon [
12].
Membrane pore blockage is mainly attributed to the accumulation of the solutes and colloids in the membrane pores and on the membrane surface, which comprises complete, internal, and intermediate pore blocking mechanisms. Membrane pore blockage, altogether with the cake filtration during the dead-end membrane filtration, was comprehensively described by Hermia’s pore-blocking models, represented elsewhere [
5,
8,
13,
14,
15].
The properties of mixed liquor play a crucial role in fouling development. The soluble microbial products (SMPs) were found to be tightly linked to the fouling intensity [
16,
17,
18]. Meanwhile, carbohydrates, which are one of the major components of the matrices of soluble microbial products in mixed liquor, were identified as the primary foulants in MBR and BF-MBR systems [
19].
Carbohydrates were found to be principally responsible for the formation of the gel and cake layer on the membrane surface, and for complete, intermediate and internal pore blocking [
20,
21,
22]. In addition, they were reported to cause concentration polarization, which entails the increased gel and cake layer thickness [
23]. Complete, intermediate and internal pore blocking are associated with irreversible and irrecoverable membrane fouling [
14,
24,
25], while gel and cake layer formation, together with concentration polarization, usually result in reversible membrane fouling [
26,
27,
28,
29]. Reversible fouling can be removed by physical cleaning, whereas irreversible fouling can only be removed by a more aggressive cleaning method―chemically enhanced backwash (CEB), cleaning in place (CIP) or “cleaning in air” (CIA). On the contrary, no techniques that could cope with the irrecoverable fouling have been developed [
11,
30]. Characteristic fouling types for the blocking mechanisms described by Hermia’s blocking filtration laws and the best suitable cleaning methods were reviewed in detail by Kulesha et al. [
8]. Based on the above-mentioned discoveries, carbohydrates are responsible for reversible, irreversible, and irrecoverable membrane fouling.
This knowledge helped to develop one of the most efficient strategies for fouling mitigation—the modification of biomass characteristics via coagulation, which has been in the center of attention of many studies [
8].
During the holistic comparative assessment of the Al- and Fe-based coagulants as membrane flux enhancers for the BF-MBR system, conducted in our previous work [
31], four different coagulants were experimentally and statistically evaluated: Prepolymerized aluminum chloride of medium and high basicity, non-prepolymerized aluminum and iron (III) sulfate. Based on the jar tests, the optimum pH values and the optimum dosage range for every selected coagulant were determined. These optimum conditions were maintained during the subsequent total recycle tests (TRT). The total recycle tests revealed that prepolymerized aluminum chloride coagulants provided the greatest extent of fouling inhibition among the studied compounds: The maximum filtration time (F) was equal to 120 min, and the maximum increase of the average normalized permeability (δ
avP
N) by 155.0–198.0% at the dose 1.1–1.9 µmolAl/mgSS. Meanwhile, prepolymerized aluminum chloride with the medium basicity exhibited a superior fouling mitigation behavior to its counterpart with the high basicity, demonstrated by a higher flux enhancing efficiency at lower dosages, and a wider range of the maximum performance of the first one.
Therefore, prepolymerized aluminum chloride with medium basicity was used during the optimization of the filtration system based on the two-level factorial design of the experiment. In contrast, non-prepolymerized aluminum and iron (III) sulfate provided 58 and 28% of the maximum increase of average normalized permeability, respectively, and 12.7 and 25 min of the maximum filtration time, respectively, which indicates a much lower extent of fouling mitigation than in the case of their prepolymerized counterparts. It is worth noting that, starting from the dose 0.5 µmolFe/mgSS during the TRT at a fixed pH, iron (III) sulfate caused the decline of the normalized permeability. The potential reason could be the tendency of iron (III) coagulant at certain concentrations to trigger the formation of the Fe-rich gel matrix of polysaccharides on the membrane surface, thus increasing reversible fouling, which is reported by several studies [
19,
32,
33]. Hence, the prepolymerized aluminum coagulant of the medium basicity is identified as the most efficient membrane flux enhancer, which entails a tenfold increase in the filtration time of the membrane separation cycle, and increases the net membrane flux by 30–56% in the BF-MBR pilot system.
Despite the extensive research work in the field of chemical flux enhancement, the processes taking place in the system after adding of the coagulants still partly remain a black box due to their complexity and rapidness [
34,
35,
36]. A deeper comprehension of the influence of the flux enhancers on the mixed liquor filterability can be attained through the investigation of the physicochemical properties of the system, which can shed some light on the interaction between the carbohydrates and the hydrolysis species of the coagulants to get a better understanding of the governing mechanisms.
Thermodynamic studies, related to the membrane fouling control in MBR, were carried out by different research teams [
37,
38,
39,
40,
41]; however, they were merely focused on the interfacial interactions between the sludge floccules and the membrane surface, and the calculation of the relevant energies according to: The XDLVO (Extended Derjaguin, Landau, Verwey, Overbeek) theory, the combined surface element integration method with the composite Simpson’s rule for the calculation of the interfacial interactions, with regard to the rough membrane surface, and the quantitative definition of the hydrophilicity/hydrophobicity of the material.
Concerning the polyaluminum hydrolysis species, the computational study of the structure of the Keggin Al
13 complex was performed by Pophristic et al. [
42]. The optimized bond lengths and angles of Al
13O
4(OH)
24(H
2O)
12Cl
7 were determined, followed by the ab initio molecular dynamics calculations. Al
13O
4(OH)
24(H
2O)
117+ was identified as the most stable configuration. Armstrong et al. [
43] studied the thermodynamic properties of the Keggin Al
13 sulfate and selenate molecules in the crystal state, in order to identify the link between their structure and their reactivity. The highly exothermic enthalpies of formation of the Keggin Al
13 clusters from the elements and oxides, and the enthalpies of the relevant 5N solutions, were reported.
The interaction between the aluminum hydrolysis species and glucose was studied by He et al. [
44], who focused on the investigation of the coordination of β-D-glucopyranose to [Al(OH)(H
2O)
n]
2+ and [Al(OH)
2(H
2O)
n]
+ ions through the Density-functional method. The formation of the double-O-ligand coordination complex was found to be thermodynamically favorable if it was formed through two O
4–O
6 adjacent oxygen atoms in the β-D-glucopyranose moiety.
However, no computational thermodynamic studies can be found on the chemical coordination of the monosaccharides to the polymeric hydrolysis species of the prepolymerized aluminum chloride, especially Al
13n+, which constitute the majority of its hydrolysis species [
45,
46] and have the highest coagulation potential [
47], thus being primarily responsible for the flux enhancing ability of the relevant coagulant.
The current work aims to fill the existing conspicuous gap in the scientific knowledge by developing a strategy, which combines the quantum chemical model and thermodynamic calculations with the multivariate chemometric approach to identify the thermodynamically favorable pathways of the formation of inorganic-organic complexes, and the thermodynamic patterns of fouling mitigation during the application of the prepolymerized aluminum coagulant of the medium basicity in BF-MBR.
2. Materials and Methods
The calculations of the thermodynamic parameters of the formation of the individual reactants and the relevant complexes were carried out, applying the semi-empirical computational molecular orbital method—Parameterized Model number 3 (PM3) using HyperChem
TM 8.0.6 software (Hypercube Inc., Waterloo, ON, Canada). PM3 is based on the neglect of diatomic differential overlap approximation. This parametrization procedure enables the acquisition of fully optimized molecular geometries and the calculation of the heats of formation, dipole moments and ionization potentials. In comparison with the MNDO (Modified Neglect of Diatomic Overlap) and AM1 model (Austin Model 1), the PM3 method is characterized by lower average absolute errors when calculating standard enthalpies, and provides more precise calculations [
48]. The PM3 method provides the enthalpies of formation with the weighted total mean absolute deviation ±10.9 kJ/mol [
49].
In order to simulate the interactions between the monosaccharides and Al13-complex, the relevant molecules were built and solvated using 216 water molecules and the periodic solvent box of the size 18.7 × 18.7 × 18.7 Å for the monosaccharides; and the periodic box of 31.3 × 31.3 × 31.3 Å using 1010 water molecules for Al13 and Al13-monosaccharide complexes. The minimum distance between the solvent and the solute atoms was kept at 2.3 Å. Full geometry optimization using a Polak-Ribiere algorithm was performed for every analyzed compound, in order to reach the conformation of the lowest potential energy. The restricted Hartree-Fock method was applied to calculate the spin interactions in every compound. The standard temperature was set in all of the simulations (298.15 K). The results of the quantum chemical calculations are reported with the accuracy of the convergence parameter, i.e., the self-consistent field energy (SCF), equal to ±0.0418 kJ/mol.
The calculation of the main thermodynamic parameters of the reactions (i.e., standard enthalpy change (Δ
Ho), standard entropy change (Δ
So), and standard Gibbs energy change (Δ
Go)) between the selected monosaccharides and Al
13 complex, resulting in the formation of the relevant Al
13-monosaccharide complexes, was performed according to Hess’s law (Equation (1)), its extension to entropy (Equation (2)) and Gibbs energy (Equation (3)), and the Gibbs-Helmholtz equation (Equation (4)).
where
is the standard enthalpy change of formation;
is the standard entropy of formation;
is the standard Gibbs energy change of formation of the individual reactants and products.
The statistical investigation of the calculated thermodynamic parameters of the studied systems was conducted using PLS regression analysis and cluster analysis. The statistical software, The Unscrambler® X10.3 (CAMO Software AS, Oslo, Norway), was used for this purpose.
3. Results and Discussion
3.1. The Main Identified Foulants
Monosaccharides (CnH2nOn, n = 3–6) and their derivatives are the main building block molecules of polysaccharides, which determine the characteristics of the latter ones. Therefore, it was decided to investigate the behavior of the monosaccharides in terms of their interaction with the selected coagulant.
Different studies investigate the presence of the monosaccharide species in the mixed liquor and the fouling layers of the membrane bioreactor (MBR) systems, as well as in the biofilm matrixes (
Table 1).
According to
Table 1, such monosaccharides as glucose, galactose, glucuronic acid, glucosamine, rhamnose, fucose, arabinose, and mannose (marked with the blue rectangle) are the most typical constituents of the polysaccharides in the mixed liquor, gel, and cake layers in the MBR systems. Since the vast majority of them were also identified in the biofilm matrices [
52], in the present study, it is assumed, that the highlighted monosaccharides make up the polysaccharides in the BF-MBR system.
Taking into account the above-mentioned references, a couple of nuances can be singled out. First, the relative proportions of the monosaccharide species in the structures of the polysaccharides in the gel and cake layers of the MBR system tend to vary, depending on the applied solids retention times (SRTs), which is demonstrated by Silva et al. [
50]. Besides, the authors point out that the variety of monosaccharides increases with the decrease of SRT and the intensification of biofouling (all of the dominant ones at different SRTs monosaccharides lie in the blue rectangle in
Table 1). Second, as reported by Miyoshi et al. [
55], certain monosaccharides might have a higher affinity to the membrane surface, and can thus cause more severe irreversible fouling than others. The clear indicator was the difference in the monosaccharide content in the fouling layer and in the mixed liquor. According to the results [
55], glucose, galactose, rhamnose and mannose were found to be dominant in the matrix of the fouling layer.
3.2. The Structure and Active Centers of the Studied Foulants
Different studies suggest the prevalence of the cyclic form (pyranose, furanose) of the selected monosaccharides (hexoses, pentose, and their derivatives) in the aqueous solutions, which can be explained by the fact that the formed cyclic hemiacetals are strain-free and more stable than the open-chain structure [
56,
57,
58,
59]. Based on the previous research works on the determination of the characteristic anomeric forms of the selected monosaccharides [
60,
61,
62,
63,
64,
65,
66,
67] and the above-mentioned findings (
Table 1), the following compounds are further considered as targeting foulants: β-D-glucopyranose (Glc), β-D-galactopyranose (Gal), β-D-mannopyranose (Man), α-D-glucopyranuronic acid (GlcUA), β-L-rhamnopyranose (Rha), β-L-fucopyranose (Fuc), α-L-arabinofuranose (Ara), and β-D-glucosamine (GlcN).
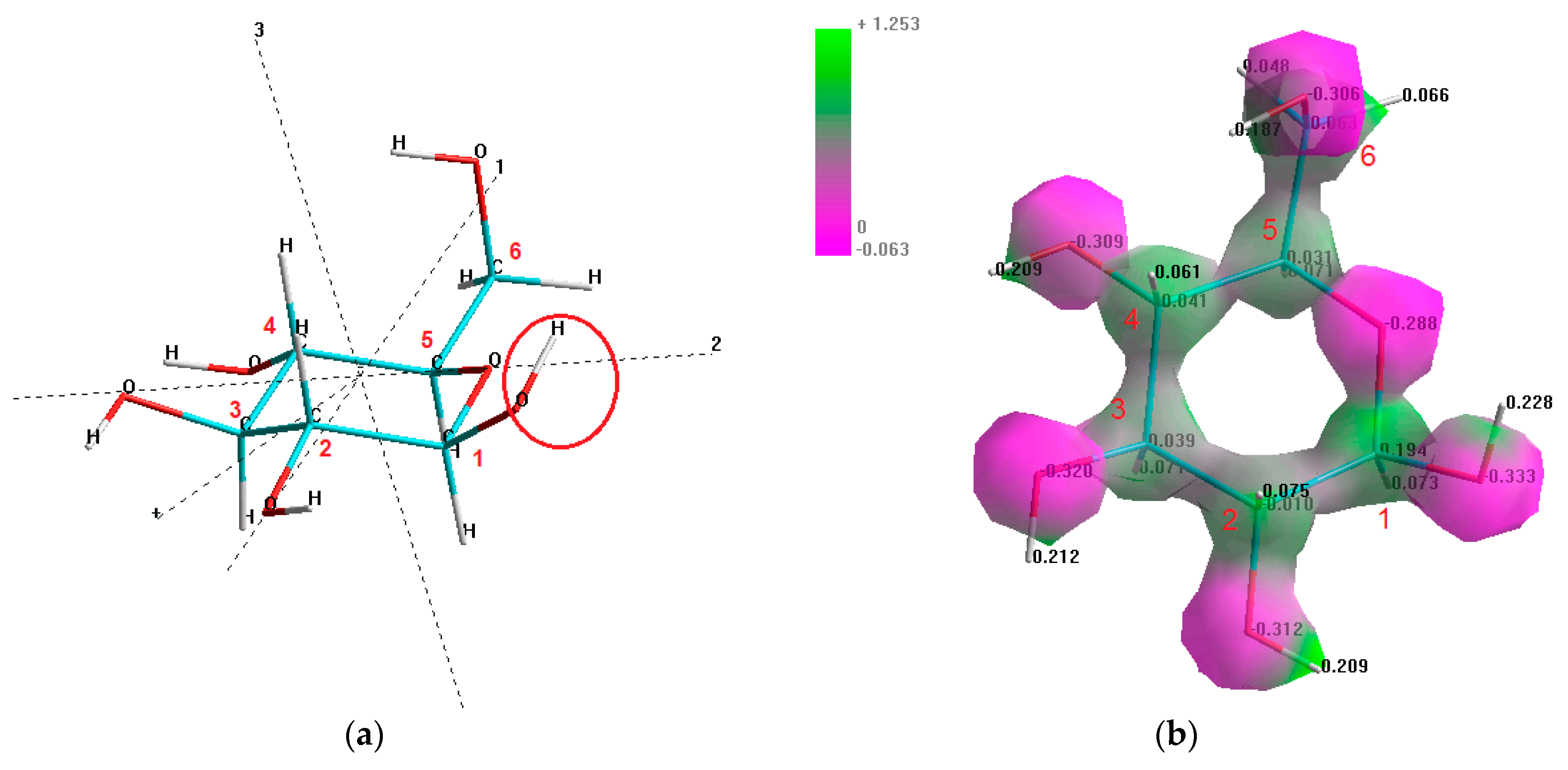
Monosaccharides in the cyclic form are characterized by an active center, which is called the glycosidic hydroxyl group. The higher reactivity of the glycosidic hydroxyl group can be explained by the influence of the ether-type oxygen atom (between C
1 and C
5) (
Figure 1a), which partially shifts the electrons from the contiguous C–O bond (in the C
1 position) to its own orbitals.
Hence, the shift of electron density increases the polarity between the C
1 carbon and the –OH group, making this hydroxyl group more chemically active [
68]. The shift of the electrons from the carbon atom in position 1 (atom C
1) to the ether-type oxygen atom in the structure of β-D-glucopyranose, is clearly indicated by the areas of the relatively higher electrostatic potential (around 1.25 e/a
0) and the increased net positive charge of C
1 (+0.194) in comparison to the other present carbon atoms, which is demonstrated in
Figure 1b.
Figure 1b demonstrates the computationally generated electrostatic potential map of the β-D-glucopyranose molecule, which visualizes the charge/electron density distribution within the molecule, represented as the isosurface. According to the gradation of color in the electrostatic potential map, the asymmetrical distribution of the electron charge density is clearly indicated in the O–H groups. The hydrogen atoms have a low electron density, and hence a net positive charge and positive electrostatic potential, as shown by the bright green color, while the oxygen atoms have a high electron density, and thus a net negative charge and negative electrostatic potential, indicated by the deep purple color. Meanwhile, the C–O groups are characterized by the accumulation of the net positive charge and positive electrostatic potential at the carbon atoms and the negative charge and negative electrostatic potential at the oxygen atoms. The accumulation of the net negative charge at the oxygen atoms can be explained by the fact that oxygen is the element of the second highest electronegativity (χ) in the periodic table (χ
O = 3.5, vs. χ
C = 2.5 vs. χ
H = 2.1 on the Pauling scale), and therefore its atoms have the highest relative ability (after fluorine) to attract the electrons of other atoms to which it is bonded [
69].
The electrostatic potential map indicates a significant part of the intermediary electrostatic potential regions, whose colors are not completely green or completely purple. This effect is explained by the nature of observed bonds in the structure of β-D-glucopyranose, as well as in the structure of the other selected monosaccharides, which is covalent polar. Meanwhile, the polarity of the O–H bonds is higher than the polarity of the C–O bonds—ΔχOH = 1.4 > ΔχCO = 1.0.
The electrostatic potential surfaces of the other investigated monosaccharides have the character similar to the one of β-D-glucopyranose and are demonstrated in
Figure S1. The β-D-glucopyranose molecule was selected for the description since it is the most stable aldohexose, and is among the most abundant monosaccharides in MBR [
50,
56].
The total dipole moment of the β-D-glucopyranose molecule is 1.639 D, with the highest contribution from the X and Z vector components (
Figure 1a).
3.3. Dominant Hydrolysis Species with Regard to the Medium Basicity Prepolymerized Aluminum Chloride
Prepolymerized aluminum coagulants are characterized by the dominance of the species with the charges higher than the charges of the majority of the species of non-prepolymerized inorganic coagulants. This feature enhances the coagulating ability of the prepolymerized aluminum coagulants and simplifies the operation process, thus making them more advantageous than their non-prepolymerized counterparts [
35]. One of the main factors which influences the dominance of certain polyaluminum hydrolysis species is the basicity of the prepolymerized aluminum coagulant [
70]. According to the previous work by Kulesha et al. [
31], prepolymerized aluminum coagulant with the medium basicity (OH/Al 1.3) exhibits the best potential to mitigate fouling in the BF-MBR system at the optimum pH range 5.5–6 (acidic), which was mainly attributed to the highest bearing charge concentration among the studied species.
Al
137+ (
Figure S2) is considered the most stable aluminum complex in the prepolymerized aluminum chloride (PACl) solution [
71,
72], which is mainly attributed to a surface positive charge with the π-electrons delocalized in the six-member (hexagon-like) ring structures [
73]. Meanwhile, the surface charge decrease makes the subsequently formed Al
137 − n species less stable [
74]. The presence of Al
137+ complexes in polyaluminum chloride solutions is the prime contributor to their efficiency enhancement, due to the increase in the charge concentration of the relevant PACl [
71,
74,
75]. This polynuclear complex as the prime component of the prepolymerized aluminum chloride solutions is first introduced and described by Johansson [
76].
The dominance of [Al
13O
4(OH)
28(H
2O)
8]
3+, (which probably originated from [Al
13O
4(OH)
24(H
2O)
12]
7+) in the medium basicity coagulant of OH/Al 1.5 at pH ≈ 3.8 at the concentration 0.1 M Al, was reported by Bottero et al. [
77], during the analysis of the nuclear magnetic resonance (NMR) spectra (~71% out of the total aluminum concentration) and computational analyses based on Glueckauf’s formula and the Debye-Hückel law (~80–88% out of the total aluminum concentration). The experimental potentiometric titration analysis and the relevant model based on its results indicated the presence of the [Al
13O
4(OH)
24(H
2O)
12]
7+ complex, which consisted of the symmetrical tetrahedrally-coordinated aluminum ion at the center of the structure with the weak/non-existent electric field gradient, surrounded by twelve octahedral aluminum ions with the relatively high electric field gradient and the potentially distorted structure.
Feng et al. [
78], who studied the speciation of different prepolymerized aluminum coagulants by applying electrospray ionization mass spectrometry, reported that the Al
133+ species was one of the main components of these prepolymerized aluminum coagulants, which was assumed to be directly transformed from Keggin-[Al
13O
4(OH)
24(H
2O)
12]
7+ complex. The coexistence of Keggin-[Al
13O
4(OH)
28]
3+ and the octahedral [Al
13(OH)
36]
3+ structures was hypothesized; meanwhile, the Keggin-Al
133+/[Al
13O
4(OH)
24(H
2O)
12]
7+ form was identified as the prevailing structure.
Pernitsky and Edzwald [
70], based on the experimental solubility data for the sulfated prepolymerized aluminum chloride coagulant of the medium basicity, hypothesize either the presence of the combination of monomers (Al(OH)
2+, Al(OH)
2+) and Al
137+, or the occurrence of some other aluminum species in the studied system at pH 5–7.
In the study by Rämö et al. [
79], who investigated the distribution of the polyaluminum species in the 1 mum Al solutions of prepolymerized aluminum chloride with the medium basicity (OH/Al 1.3), based on the mass spectrometry (MS) analysis, the dominance of Al
132+ and Al
133+ compounds, which made up 72% out of the total ion count at pH 4.7, is reported. No detection of [Al
13O
4(OH)
24(H
2O)
12]
7+ could be explained by any availability of the counter anions in the system and the drying droplet of the specimen. Some larger formations, for example, Al
26, were also observed; however, the Al
13 complex is the dominant species in the system. On the contrary, the monomers, dimers, and trimers of aluminum were completely absent [
46].
According to mass spectrometric studies, reported by Sarpola [
45], the PACl with the medium basicity in the pH range 4.65–6.46 is characterized by the following protonated open form of Al
13—[Al
13O
4(OH)
29]
2+. Due to the high stability of the [Al
13O
4(OH)
24(H
2O)
12]
7+ complex, it is suggested that the complex in its usual form is less likely to participate in any reactions. Meanwhile, it is hypothesized [
46] that after the formation of [Al
13O
4(OH)
24(H
2O)
12]
7+ in the aqueous solutions, the four-coordinate oxygen atoms in each ring, which are shared with the central aluminate (
Figure S2), get “over-bonded”, and thus “loose” the bond with one of the other twelve aluminum atoms that they are connected to, getting protonated and exposing the open chains or ring substructures to water. Hence, four rings with the asymmetrical charge division are produced, giving rise to the active centers, which can potentially attract the negatively charged systems and react with them [
46].
Seichter et al. [
80], who investigated the species of prepolymerized aluminum chloride, formed by hydrolysis and condensation, assigns the detected complex cations to the octahedral structure [Al
13(OH)
24(H
2O)
24]
15+, which is identified as the other important tridecameric cation, in addition to the Keggin (tetrahedral) type. This complex, as the complex [Al
13O
4(OH)
29]
2+ introduced by Sarpola [
45], has the planar core, and thus is potentially much more likely to participate in reactions with the organic matter, as well as undergo further polymerization. However, the formation of this polycation structure was expected to occur autonomously from the Keggin-type cation.
Based on the discoveries by Sarpola [
45] and all above-mentioned findings, the present study assumes that during coagulation, applying the prepolymerized aluminum chloride with medium basicity, the open structure of Al
13 complex in the form of [Al
13O
4(OH)
29]
2+ plays the most crucial role in the reactions with the oppositely charged foulants.
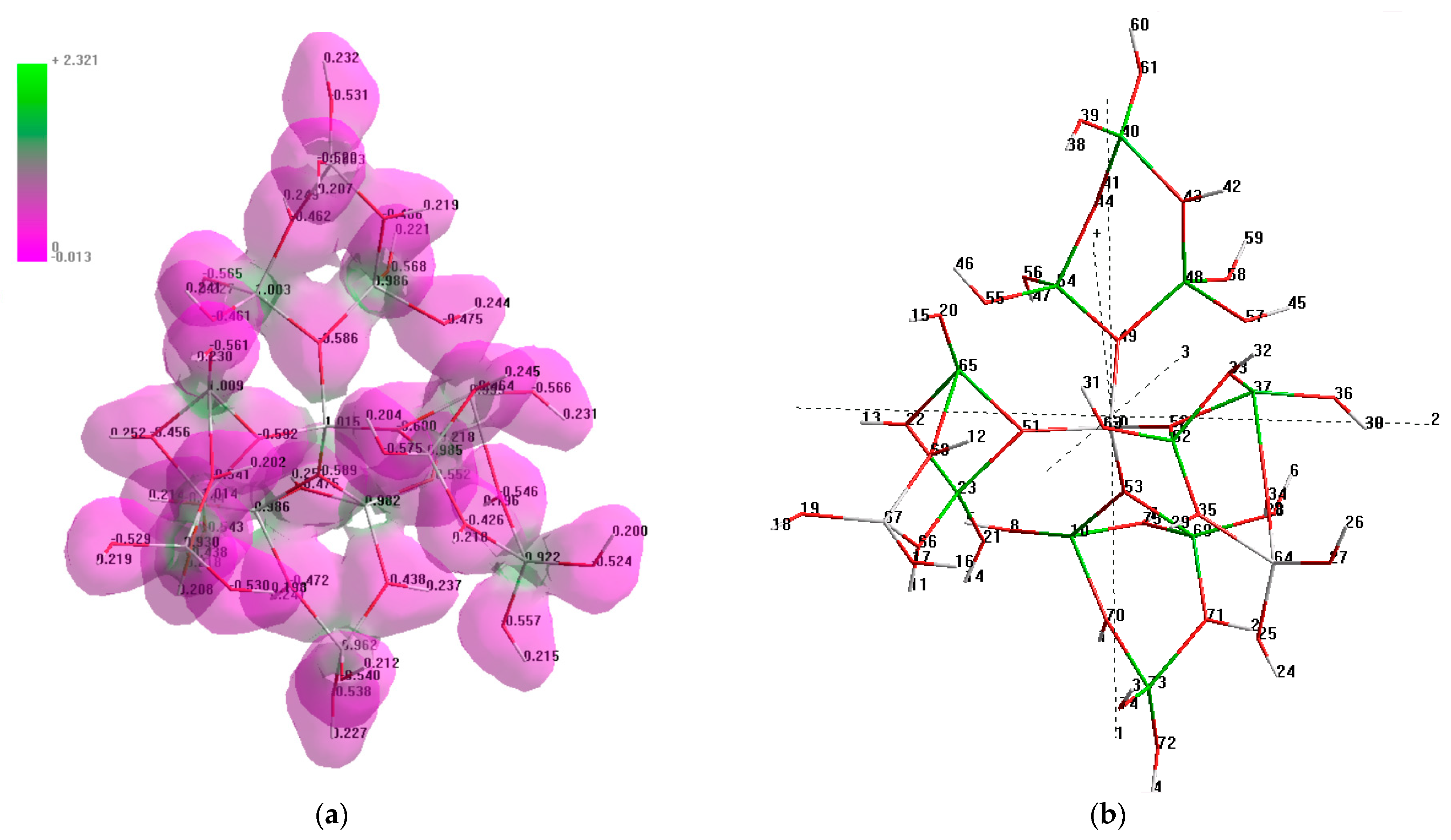
3.4. The Structure of Al132+
The planar open structure of the Al
133+ complex was introduced by Sarpola [
45], and taken as the basis for building Al
132+. Meanwhile, the geometry optimization analysis conducted in the present work indicates that the minimum of potential energy can only be reached if the molecule has a non-planar conformation, as represented in
Figure 2.
According to the charge/electron density distribution within the molecule, represented as the isosurface of the Al
13-complex (
Figure 2a), aluminum atoms have a low electron density, a net positive charge and a positive electrostatic potential, as shown by the bright green color; meanwhile, the oxygen atoms have a high electron density, and thus a net negative charge and a negative electrostatic potential, indicated by the bright purple color. According to the represented electrostatic potential map, all aluminum atoms, except for the central tetrameric aluminum, are the potential active centers during the interaction with the foulants. Meanwhile, based on the charge balance calculations, it was identified that Al no. 64 (
Figure 2b) has a slight net negative charge (−0.023), and that Al no. 67 has almost no charge (0.004); thus, both are less likely to participate in these reactions. Consequently, the aluminum atoms, highlighted with the light green color in
Figure 2b, are the potential active centers of the Al
13-complex. The total dipole moment of the Al
13 complex is 7.25 D. The analysis of the components of the total dipole moment demonstrates the maximum contribution from the Z vector component (
Figure 2b).
The nature of the observed bonds in the structure of the Al13-complex is ionic, since the polarity of the O–Al bonds (ΔχAlO) is equal to 2 (χO = 3.5, χAl = 1.5 on the Pauling scale). The great difference in electronegativities is also indicated by the significant differences in the electron density at the aluminum and oxygen atoms, which is demonstrated by the entirely green and entirely purple regions, respectively, with no regions of intermediary electrostatic potential.
3.5. Results of the Quantum Chemical Simulations and Calculations
It was decided to investigate the Al
13-monosaccharide complex formation through the double-O-ligand coordination, since the formed structure would contain an additional ring system with the delocalized π-electrons that would potentially contribute to its stabilization. Based on the findings described in
Section 3.2, and the structural peculiarities of the selected monosaccharides, the formation of the Al
13-monosaccharide complexes can occur through the following pairs of the carbon atoms with the adjacent oxygen atoms:
- (1)
C1–C2, C1–C6, C4–C6, C1–C3 for the aldoses and uronic acid (β-D-glucopyranose, β-D-galactopyranose, β-D-mannopyranose, and α-D-glucopyranuronic acid);
- (2)
C1–C2, C1–C4, C2–C3, C1–C3 for the deoxy sugars (β-L-rhamnopyranose and β-L-fucopyranose);
- (3)
C1–C2, C2–C3, C3–C5, C1–C3 for the pentose (α-L-arabinofuranose);
- (4)
C1–C6, C1–C4, C4–C6, C1–C3 for the aminosugar (β-D-glucosamine).
The pairs of aluminum atoms in the Al
132+ complex, which can potentially participate in the interaction with the listed active centers of the monosaccharides, are specified in
Section 3.4.
It was assumed that the relevant active centers of the monosaccharides first got deprotonated, i.e., ionized, and then they participated in the chemical coordination with the Al
13 complex. As a result of these interactions, two water molecules are dehydrated, which was taken into consideration during the relevant calculations. The enthalpy and Gibbs energy of formation of the liquid water molecule specified by Dean [
81] were used in this study. The entropy change for water molecules was calculated from the reference data [
81] according to Equation (2).
The assessment of the spontaneity of the potential interactions between the selected monosaccharides and Al132+ complex was performed through two steps: First, a series of quantum chemical simulations were conducted, focused on the determination of the thermodynamic parameters of the formation of the individual reactants and the relevant complexes—standard enthalpy change (ΔHof), standard entropy (Sof), and standard Gibbs energy change (ΔGof); Second, the calculation of the main thermodynamic parameters of the reaction between the selected monosaccharides and Al132+ complex, i.e., the process of the formation of the Al13-monosaccharide complex—standard enthalpy change (ΔHo), standard entropy change (ΔSo), and standard Gibbs energy change (ΔGo) was performed according to Equations (1)–(4).
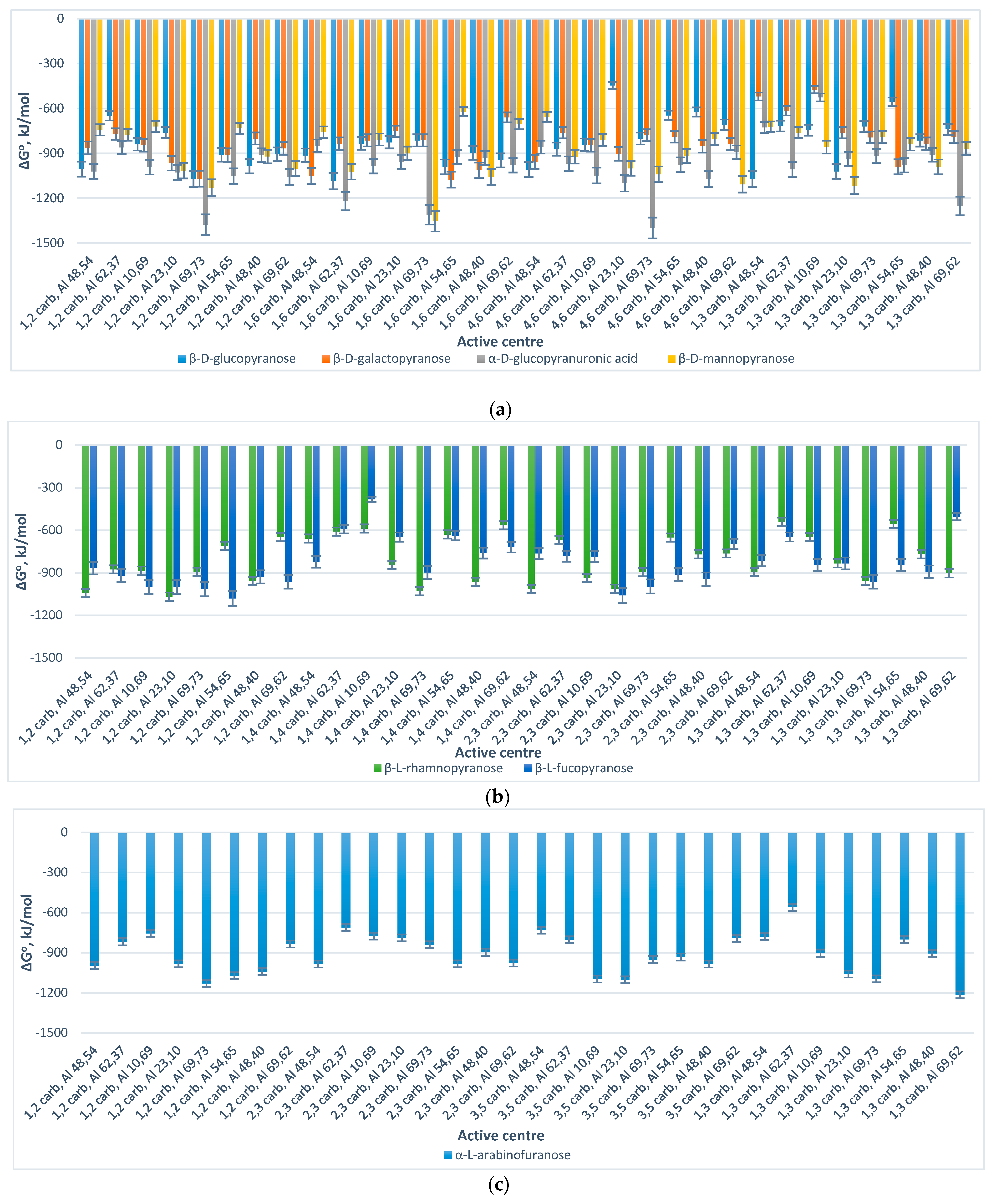
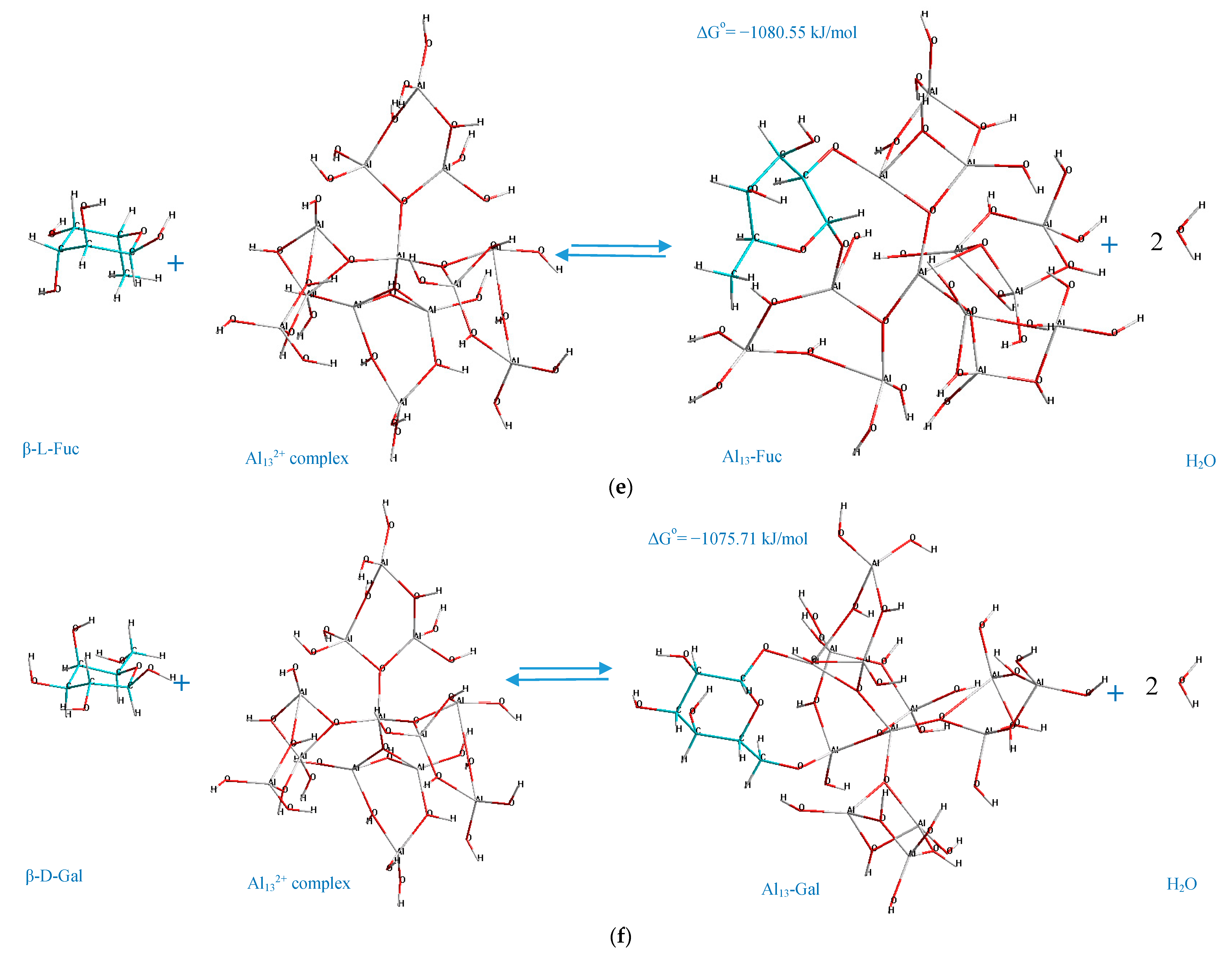
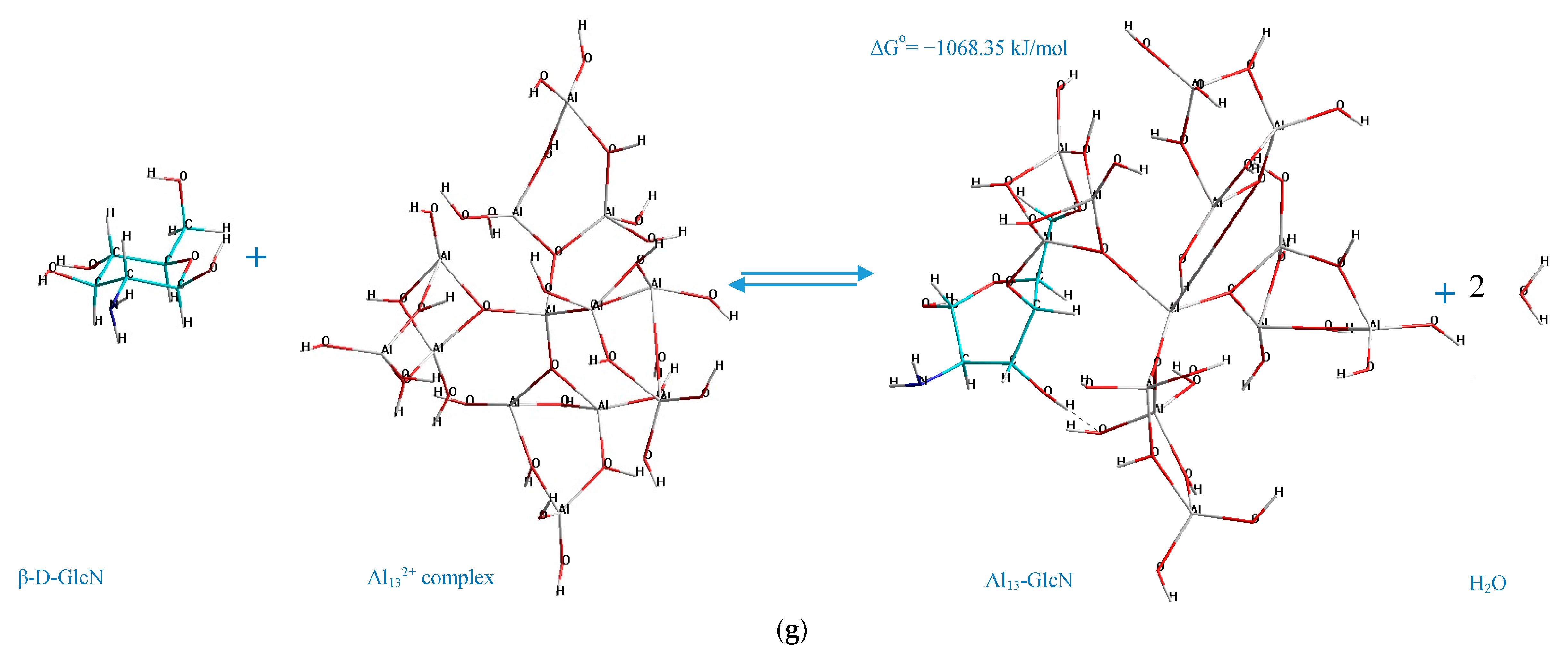
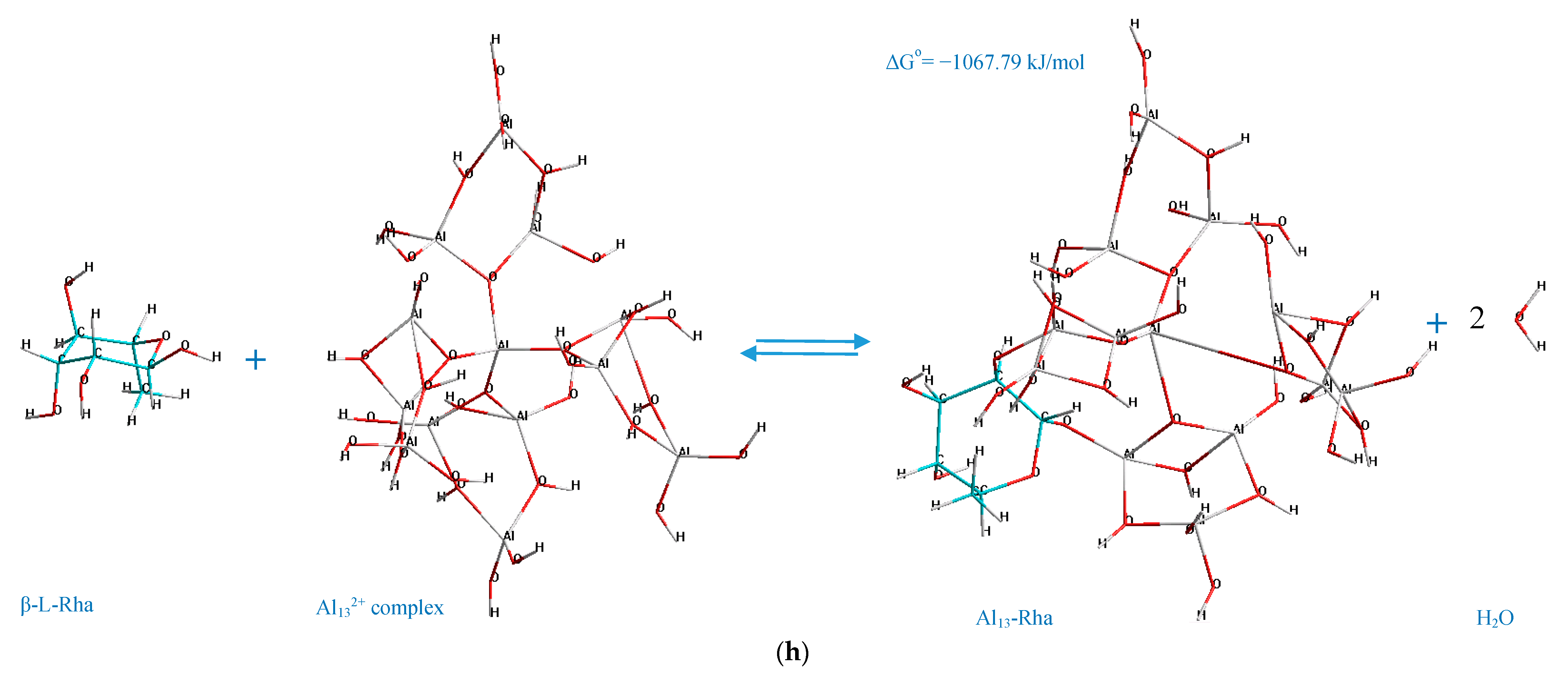
The standard Gibbs energy change of the process of formation of Al
13-monosaccharide complex (interaction between the selected monosaccharides and Al
132+ complex), which is the main indicator of the spontaneity in chemical reactions, is represented in
Figure 3.
The analysis of the acquired charts (
Figure 3) demonstrates that all of the processes which result in the formation of the suggested Al
13-monosaccharide complexes have a negative standard Gibbs energy, which indicates that the processes of their formation are spontaneous, and hence thermodynamically favorable. Meanwhile, the thermodynamic stability of the formed Al
13-monosaccharide complex highly depends upon the active centers of the monosaccharides and the Al
132+ complex, which participate in the chemical coordination process, and the nature of the monosaccharide. The following complexes are identified as the most thermodynamically stable, since they exhibit the highest negative values (with regard to every considered monosaccharide as the reactant) of standard Gibbs energy change: 4,6 GlcUA, Al 69,73 (Δ
Go = −1398.87 kJ/mol); 1,6 Glc, Al 62,37 (Δ
Go = −1085.82 kJ/mol); 1,6 Gal, Al 54,65 (Δ
Go = −1075.71 kJ/mol); 1,6 Man, Al 69,73 (Δ
Go = −1353.75 kJ/mol); 1,2 Rha, Al 23,10 (Δ
Go = −1067.79 kJ/mol); 1,2 Fuc, Al 54,65 (Δ
Go = −1080.55 kJ/mol); 1,3 Ara, Al 69,62 (Δ
Go = −1215.10 kJ/mol); 4,6 GlcN, Al 69,73 (Δ
Go = −1068.35 kJ/mol).
With regard to the general tendencies, the aldoses and uronic acid exhibit similarly high (−1376.16–(−1069.30) kJ/mol) negative values of the Gibbs energy change of the interaction between the oxygen atoms at the C
1–C
2 active centers of the monosaccharide skeleton and the aluminum atoms of the Al
132+ complex no. 69, 73 (
Figure 3a).
Meanwhile, the deoxy sugars demonstrated similarly high (−1067.79–(−998.74) kJ/mol) negative values of the Gibbs energy change for the interaction process between the oxygen atoms at C
1–C
2/C
2–C
3 atoms of the monosaccharide skeleton and the aluminum atoms of the Al
132+ complex no. 23, 10 (
Figure 3b).
Apart from 1,3 Ara, Al 69,62 complex, the pentose, represented by α-L-arabinofuranose, showed relatively high negative values of the Gibbs energy change of the process of formation of the Al
13-monosaccharide complexes through: (1) C
1–C
3/C
1–C
2 atoms of the monosaccharide and the Al atoms of the Al
132+ complex no. 69, 73 (−1129.30–(−1094.84) kJ/mol); (2) C
1–C
3/C
3–C
5 atoms of the monosaccharide and the Al atoms of the Al
132+ complex no. 23, 10 (−1102.18–(−1059.07) kJ/mol); (3) C
3–C
5 atoms of the monosaccharide and the Al atoms of the Al
132+ complex no. 10, 69 (−1096.97) kJ/mol) (
Figure 3c).
Concerning the aminosugar, represented by β-D-glucosamine (
Figure 3d), considerably high negative values of the Gibbs energy change of the process of interaction of the oxygen atoms at the C
1–C
3 atoms of the aminosugar and Al atoms of the Al
132+ complex no. 54, 65; and the oxygen atoms at the C
4–C
6 atoms of the aminosugar and Al atoms of the Al
132+ complex no. 10, 69 were obtained (−1057.95–(−1036.44) kJ/mol).
The standard entropy change and enthalpy change of the processes resulting in the formation of the investigated Al
13-monosaccharide complexes is demonstrated in
Figures S3 and S4. According to the results (
Figure S3), the standard entropy change is negative for all the processes, which can be explained by the fact that the spontaneous association of such reactants as monosaccharide and the Al
132+ complex gives the rise to a more compact/organized structure [
82].
Concerning the standard enthalpy change of the interaction between the monosaccharides and the Al
132+ complex, it is highly negative with regard to every investigated process, which results in the formation of Al
13-monosaccharide complex (
Figure S4).
According to Equation (4), the most favorable condition for the formation of any compound is established if Δ
H < 0 and Δ
S > 0, which indicates that this process of formation can occur spontaneously at any given temperature. As shown above, the Δ
So of the processes of formation of all of Al
13-monosaccharide complexes is negative, and the processes of their formation are exothermic (Δ
Ho < 0). In this case, there is a competition between the entropy (the level of the disorder of the system) and enthalpy factors: The first parameter facilitates the reversible process (complex decomposition), while the latter one favors the forward reaction (complex formation) [
83]. High negative values of Δ
Go for all investigated processes result from the dominance of the enthalpy factor at the standard temperature, meaning that this temperature (T = 298.15 K) is low enough to facilitate the formation of the Al
13-monosaccharide complex (T < Δ
Ho/Δ
So). However, at very high temperatures (T > Δ
Ho/Δ
So), the process of complex formation will not occur spontaneously.
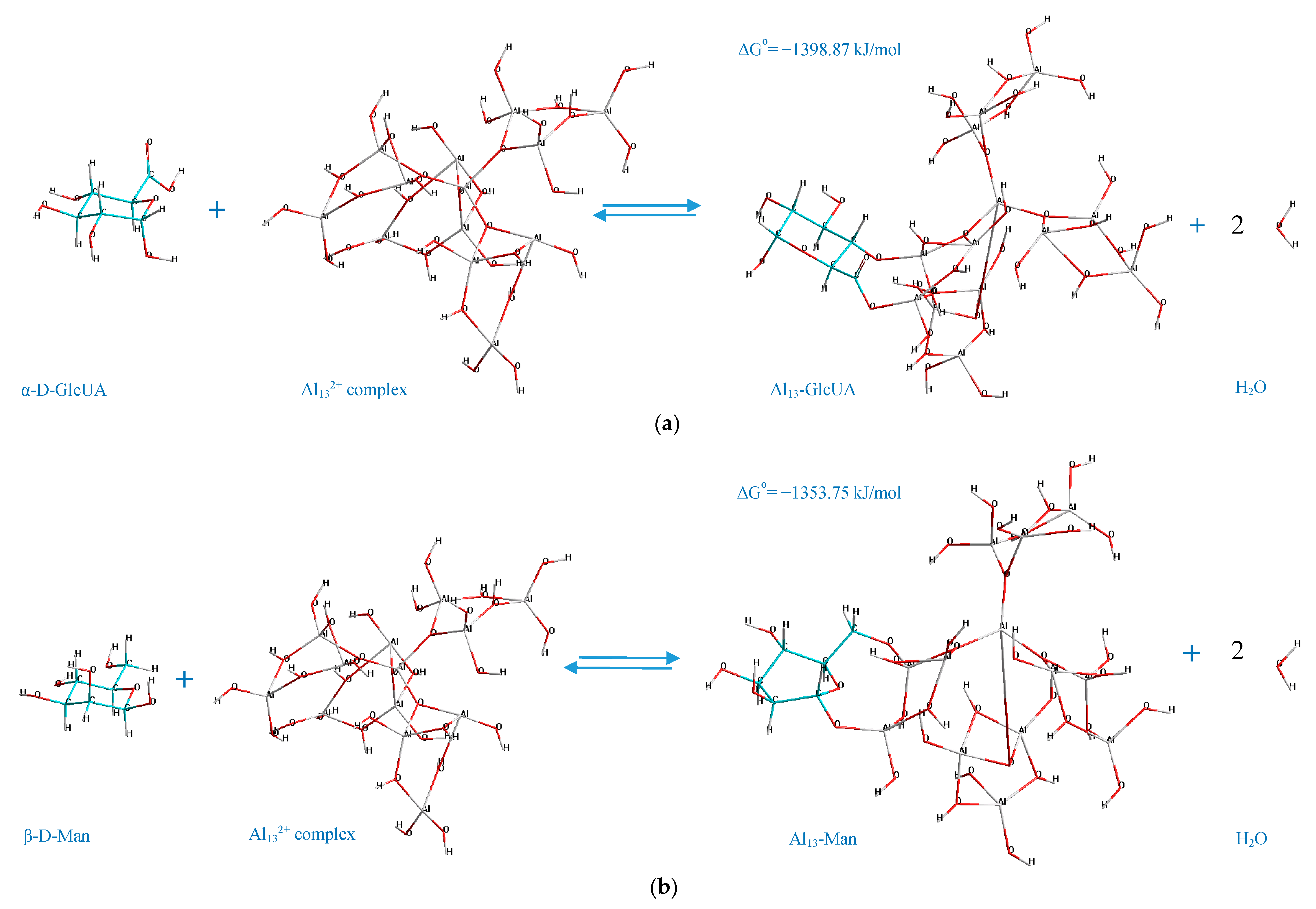
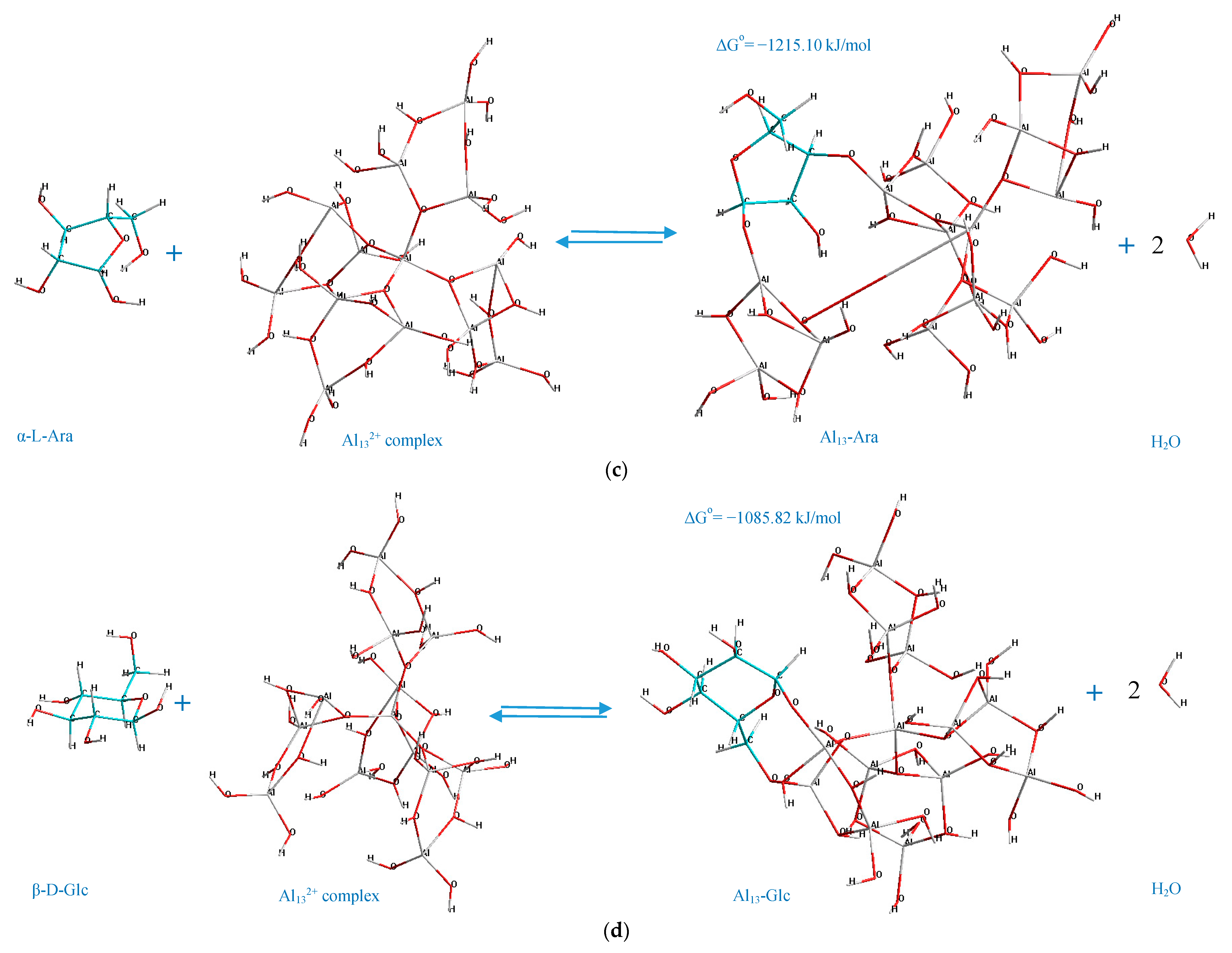
3.6. Mechanisms of the Interactions
Based on the acquired results, the optimized geometric structures of the most thermodynamically stable Al
13-monosaccharide complexes (with respect to every single monosaccharide considered as the reactant), and the determined mechanisms of their formation during flux enhancement in BF-MBR, applying prepolymerized aluminum chloride with the medium basicity, are demonstrated in
Figure 4.
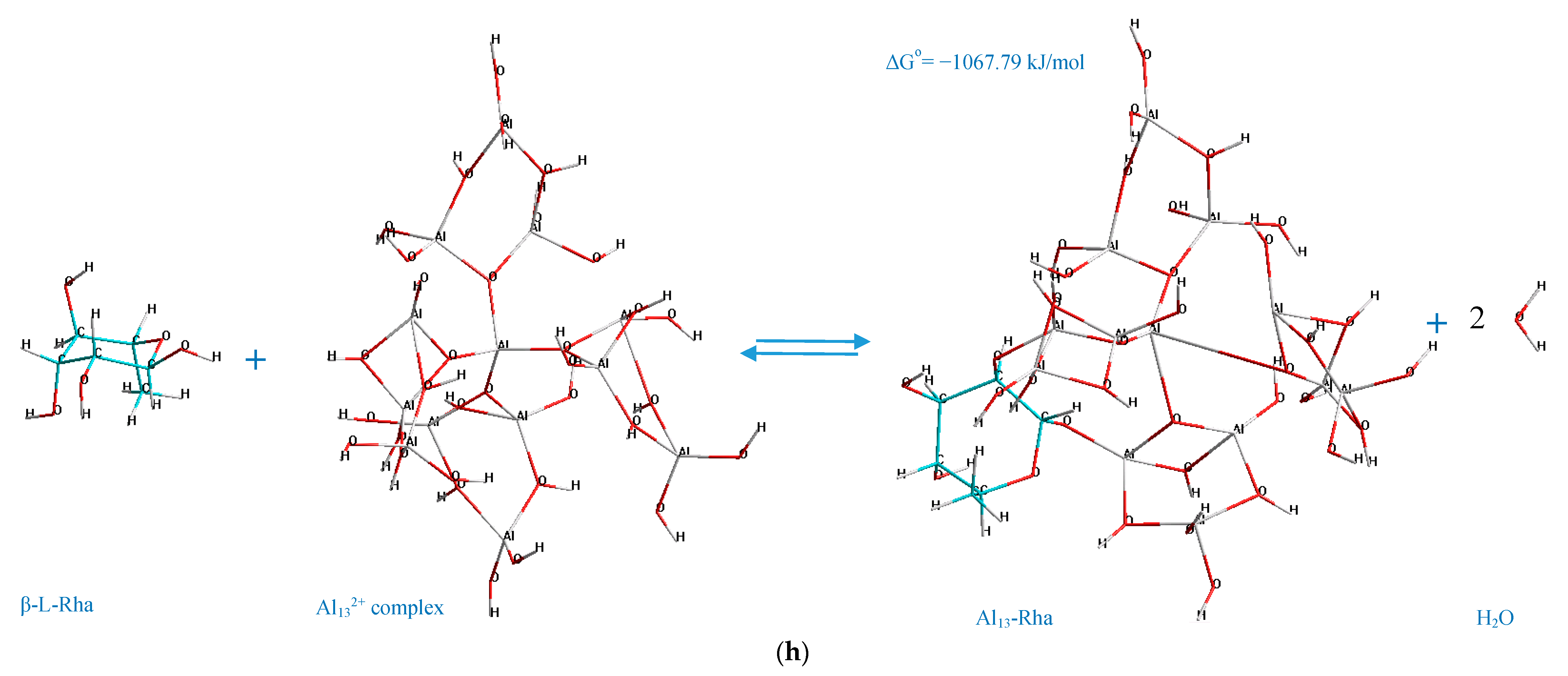
According to the determined mechanisms (
Figure 4), the most thermodynamically stable complex (Δ
Go = −1398.87 kJ/mol) is formed when the atoms of aluminum no. 69, 73 interact with the oxygen atoms in the positions C
4–C
6 of the α-D-glucopyranuronic acid (complex 4,6 GlcUA, Al 69,73). The least thermodynamically stable complex in the selected group (Δ
Go = −1067.79 kJ/mol) is formed during the interaction of aluminum atoms no. 23, 10 of the Al
13-complex and the oxygen atoms of β-L-rhamnopyranose in the positions C
1–C
2 (complex 1,2 Rha, Al 23,10).
Hence, the ranking trend among the studied complexes in decreasing order of thermodynamic stability can be classified as: Al13-GlcUA > Al13-Man > Al13-Ara > Al13-Glc > Al13-Fuc > Al13-Gal > Al13-GlcN > Al13-Rha.
The double-O-ligand coordination results in the dehydration of two H2O molecules to comply with the requirement of four-coordinate Al atoms in Al132+ complex, considering two O-ligands in the monosaccharide molecule, which was taken into account during the determination of the standard Gibbs energy change of the process of the formation of every represented Al13-monosaccharide complex.
The potential factors, which facilitate the described mechanisms of the interaction between the monosaccharides and Al
13-complex (
Figure 4) by positively contributing to the stabilization of the Al
13-monosaccharide coordinated complexes, can be: The steric effects and the polarization of Al active centers towards the O
i–O
n atoms of the monosaccharide. The polarization of the Al
n+ center in the Al
13 complex towards the O-ligand in the structure of the monosaccharide can originate from the transfer of the electrons from the 2p orbital of oxygen to the empty 3S/3p orbital of the Al ion (donor-acceptor mechanism), which, according to He et al. [
44], has the major influence on the stabilization of the coordinated complexes, formed during the double-O-ligand coordination of β-D-glucopyranose to [Al(OH)(H
2O)
4]
2+. Concerning the intramolecular hydrogen bonding, in the present work, it was only observed in the case of the complex formed by the C
4–C
6 atoms of β-D-glucosamine and Al no. 69, 73 of the Al
13 complex (the hydrogen-bonding interaction of the type O–H···O, where the O–H group is connected to the C
3 atom of the β-D-GlcN and the O atom is connected to Al no. 48 of the Al
13 complex, demonstrated by the dashed line in
Figure 4g). The other represented complexes did not contain the newly formed intramolecular hydrogen bonds; thus, this factor was excluded as the major contributor to the stabilization of the investigated complexes. However, the role of the stabilizing factors in the formation of Al
13-monosaccharide complexes should be the subject of a further investigation.
3.7. Statistical Analysis
Partial least squares analysis (PLS) of the thermodynamic parameters acquired during all simulations was conducted according to the following variables and response function (
Table 2).
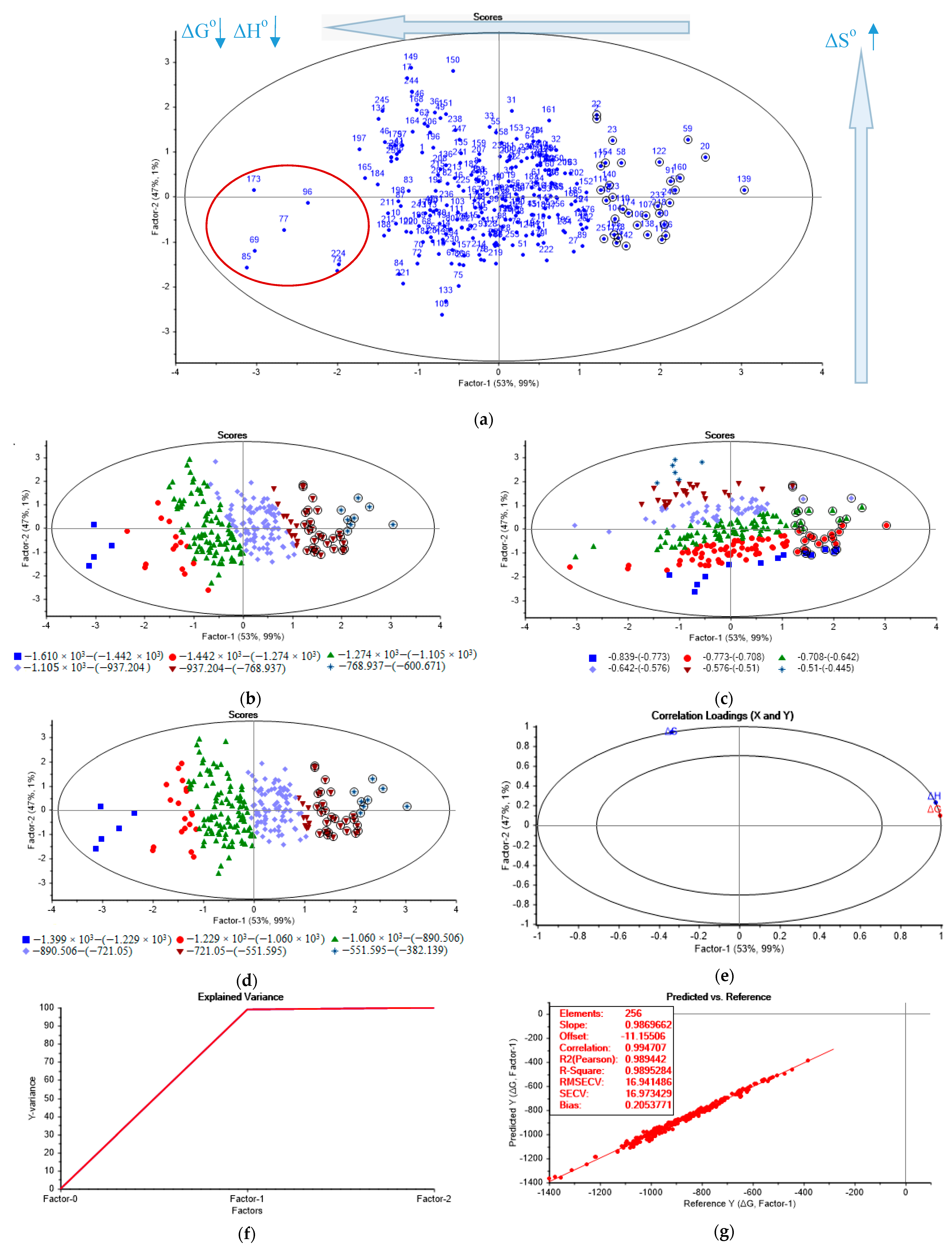
After the weighting at the first stage of the analysis to reduce the inherent differences of two predictor variables having the different ranges, the obtained model underwent random cross-validation in PLS, during which the dataset was divided into 20 segments, with 12–13 samples in each segment. The number of PLS factors was chosen according to the explained variance.
The output from the PLS analysis with regard to the scores and loadings plots is demonstrated in
Figure 5.
According to
Figure 5, the first two factors in total describe 100% of the variance in the dataset for x and y.
The scores plot demonstrates no clear clusters of similar samples (
Figure S5), which can be explained by a large number of category variables. Meanwhile, the samples, marked with the dark red oval, which are located leftmost, in the third quadrant of the scores plot (
Figure 5a), exhibit the highest negative standard enthalpy change (Δ
Ho) and Gibbs energy change (Δ
Go)—−1610–(−1442) kJ/mol and −1399–(−1229) kJ/mol, respectively, which was indicated by the results of the data-driven sample grouping (
Figure 5b,d) and the correlation loadings plot (
Figure 5e). The data-driven sample grouping was automatically conducted by equally dividing the band of the target thermodynamic parameter values in the whole range between the upper and lower limits into six groups, shown in
Figure 5b–d. The lower limits of values for the standard enthalpy change and Gibbs energy change—−1610–(−1442) kJ/mol and −1399–(−1229) kJ/mol, respectively, were selected as the targeting levels of interest, since they indicate the most thermodynamically stable complexes [
82]. The samples, marked with the dark red oval in
Figure 5a, designate the complexes with the highest negative values of the standard Gibbs energy change and enthalpy change, formed by the interaction of:
- (1)
O atoms at C1C6 atoms of β-D-Man with the Al atoms no. 69, 73 of the Al132+ complex (sample no. 173, ΔGo = −1353.75 kJ/mol, ΔHo = −1529.72 kJ/mol);
- (2)
O atoms at C1C3 atoms of α-D-GlcUA with the Al atoms no. 69, 62 of the Al132+ complex (sample no. 96, ΔGo = −1251.41 kJ/mol, ΔHo = −1437.80 kJ/mol);
- (3)
O atoms at C1C6 atoms of α-D-GlcUA with the Al atoms no. 69, 73 of the Al132+ complex (sample no. 77, ΔGo = −1310.25 kJ/mol, ΔHo = −1507.25 kJ/mol);
- (4)
O atoms at C1C2 atoms of α-D-GlcUA with the Al atoms no. 69, 73 of the Al132+ complex (sample no. 69, ΔGo = −1376.16 kJ/mol, ΔHo = −1580.16 kJ/mol);
- (5)
O atoms at C4C6 atoms of α-D-GlcUA with the Al atoms no. 69, 73 of the Al132+ complex (sample no. 85, ΔGo = −1398.87 kJ/mol, ΔHo = −1610.27 kJ/mol);
- (6)
O atoms at C1C6 atoms of α-D-GlcUA with the Al atoms no. 62, 37 of the Al132+ complex (sample no. 74, ΔGo = −1220.22 kJ/mol, ΔHo = −1440.80 kJ/mol);
- (7)
O atoms at C1C3 atoms of α-L-Ara with the Al atoms no. 69, 62 of the Al132+ complex (sample no. 224, ΔGo = −1215.10 kJ/mol, ΔHo = −1432.96 kJ/mol).
Consequently, the complexes formed by α-D-glucopyranuronic acid and the Al atoms no. 69, 73; 62, 37; and 69, 62 of the Al132+ complex make up the majority of the most thermodynamically stable samples.
According to the PLS correlation loadings plot (
Figure 5e), Factor-1 clearly accounts for the standard enthalpy change (Δ
Ho) and the Gibbs energy change (Δ
Go), while Factor-2 mainly describes the standard entropy change (Δ
So). All of the variables explain more than 50% of the variance, and hence have high importance in relation to Factor-1 and Factor-2. Δ
Ho has a high positive correlation with Δ
Go and both are negatively correlated with Δ
So along Factor-1.
The explained variance plot (
Figure 5f) indicates that the optimum number of factors is one, which provides 98.9% of the explained Y-variance. The increase in the number of factors does not significantly influence the explained variance, which was expected due to a relatively small number of variables (i.e., two variables result in two factors at most).
The analysis of the validation plot (
Figure 5g) indicates that the developed model has a linear trend (R-squared = 0.99) with a good fit for the majority of the data (slope = 0.987). The model is reliable, since the value of R-squared (Pearson) is close to the R-squared correlation (0.989 vs. 0.995). The Root Mean Square Error of Cross Validation (RMSECV) and the standard error of cross-validation (SECV) are equal to 16.94 and 16.97 kJ/mol, respectively. The obtained bias of the model is insignificant (0.21), which indicates its low tendency to any over- or underestimation of the validation values.
The stability and applicability limit of the partial least squares regression (PLSR) model was checked through the case scenarios based on the addition of the proportional and additive noise, i.e., the noise which typically affects instrument amplification and the measurement signal, respectively, to both the predictors and response function, according to Equation (5) [
84]:
where
PN is the level of proportional noise, %;
AN is the level of additive noise,
where
P% is the level of approximate additive noise in percent;
N(m,s) is the randomly distributed value, where m—mean standard deviation and s—standard deviation;
M(
i,
k)—the real raw value, acquired during the quantum chemical and thermodynamic calculations;
Mnew(
i,
k)—the value with the added tested noise.
According to the performed series of calculations, the derived partial least squares regression (PLSR) model is applicable (R-squared = 0.7) for the maximum proportional noise equal to 6% and the maximum standard deviation for the additive noise equal to 14, applied for both of the predictors (ΔHo, ΔSo) and the response function (ΔGo).
Due to the scattered nature of the scores, i.e., the lack of indication of distinctive clusters, it was decided to conduct the cluster analysis of the scores to understand if any grouping of the samples can be performed based on their similarities with regard to the specified variables.
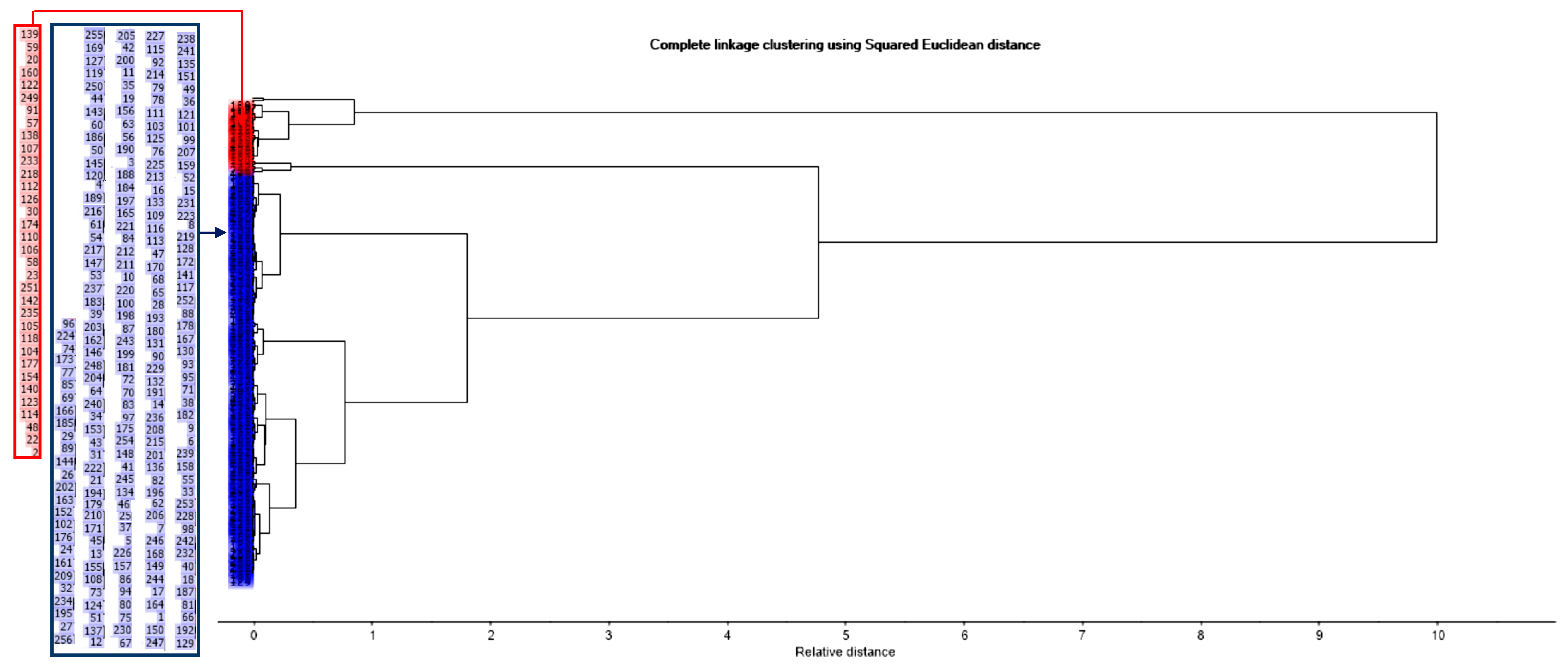
The Y-scores for the latent variables Factor-1 and Factor-2 acquired during the PLSR analysis were used as the input data for the cluster analysis. This was done in order to screen out the noise of the raw data and use the prevailing differences of the most and least thermodynamically stable complexes. The classes were generated from the scores by applying a hierarchical complete-linkage clustering method and the squared Euclidean distance as the dissimilarity function.
The acquired dendrogram visualizes the clustering (
Figure 6), which corresponds to the Y-scores in the four quadrants of
Figure 5a–d. Six classes can be identified at a higher resolution (a relative distance of around 0.8) (
Figure 6), which correspond to the six groups of the standard Gibbs energy change in the scores plot, generated during the data-driven sample grouping (
Figure 5d).
Meanwhile, at a lower resolution (a relative distance of around 4.9), merely two classes can be identified (
Figure 6). The red cluster is characterized by the samples with high values of Factor-1 and generally low values of Factor-2. The corresponding samples were marked with the black circles in
Figure 5a–d. This position of the marked samples on the scores plot indicates that they are characterized by the low negative values of the standard enthalpy change (Δ
Ho) and Gibbs energy change (Δ
Go) and the predominantly high negative values of the standard entropy change (Δ
So). Thus, the relevant complexes are less thermodynamically stable. The blue cluster represents the scores with low values of Factor-1, i.e., the corresponding samples are characterized by the relatively high negative values of Δ
Ho and Δ
Go (all of the samples which do not have the black-circle mark in
Figure 5a–d). Hence, the corresponding complexes are more thermodynamically stable. The location of the scores of the certain sequence numbers, which constitute each cluster shown in the dendrogram, can be found in the scores plot with no sample grouping (
Figure 5a).
The described clusters, generated by cluster analysis, represented the Al-monosaccharide complexes, which are more or less liable to decompose during coagulation and can be assigned to the categories “Less stable” and “Stable”, respectively.
3.8. The Working Hypothesis, Implication of the Findings, Challenges, and Perspectives
Systematic research, conducted in the present work, develops the concept of the targeting chemical influence of the Al-based prepolymerized coagulant on the fouling propensity of mixed liquor in the MBR and BF-MBR systems. Particularly, this study focuses on the investigation of the thermodynamic patterns and the mechanisms of the removal of the carbohydrates, which were proven to be the major foulants, deteriorating the filtration performance of MBR and BF-MBR.
The need for this approach was emphasized by the latest work of this research team [
31], which, as well as most of the other previous studies, dealt with the investigation of mechanisms of fouling mitigation via coagulation in MBR and BF-MBR in relation to the substrate, with no focus on the chemical fouling mitigation based on the carbohydrate composition [
85,
86,
87].
Based on the analysis of zeta potential profiles and the intrinsic coagulant charges, Kulesha et al. [
31] found out that the principal mechanism of chemical flux enhancement applying inorganic coagulants in BF-MBR is adsorption/charge neutralization. According to Stumm and Morgan [
88], the charge neutralizing process accompanies the fixation of the multivalent cations onto the deprotonated group of macromolecular sol (carbohydrates, proteins and their derivatives). This fixation is an electrostatic or a chemical interaction. Meanwhile, the chemical coordination is of considerable significance in coagulation and flocculation reactions between multivalent cations and the macromolecular sols, producing soluble and insoluble complexes, which may have various extents of cross-linkage [
88].
The underlying hypothesis of the present research is that purely chemical factors must be considered, apart from the electrostatic double-layer interactions, in order to comprehensively explain the coagulation processes occurring during the addition of the chemical flux enhancers in BF-MBR [
88]. No studies on MBR/BF-MBR, which proved/refuted this hypothesis, are found.
The current study demonstrates that the investigation of the chemical coordination processes is among the key approaches to the comprehension of the fouling mitigation mechanisms in BF-MBR. The results on the reduction of the content of soluble microbial products (SMPs) after chemical dosing, acquired in our previous work [
31], as well as in the studies by the other research teams [
27,
85,
89], can be satisfactorily interpreted with regard to chemical interactions, described in the current work, while the electrosurface phenomena, described by the theories of double layer and electrostatic interactions, are not applicable to the explanation of the interactions between the solutes/macromolecular sols and the soluble hydrolyzed coagulant species.
According to Wu and Huang [
90], SMPs exhibit a higher contribution to membrane fouling in MBR than the colloidal matter. Meanwhile, carbohydrates were found to be the dominant species in SMPs [
91]. The carbohydrate fraction of the SMPs highly correlates with fouling in contrast to the protein fraction.
Specifically, carbohydrates were found to be responsible for the reversible, irreversible, and irrecoverable fouling, while the proteins demonstrated occasional or no quantitative relationship with the fouling extent [
34,
90,
92,
93,
94]. On the other hand, in the studies by Choi and Ng [
95] and Zhou et al. [
96], the proteins exhibited a high fouling propensity in the submerged MBR. Hence, the role of the proteins in membrane fouling still remains controversial, and needs further investigation.
The general mechanisms of fouling mitigation in BF-MBR were proven to be almost the same as in the MBR systems [
31], which allows for assuming that the investigated content of the mixed liquor in MBR, represented by different research groups [
50,
51,
53,
54,
55], is also valid for the BF-MBR.
Due to the lack of the reference data on the thermodynamic parameters of formation of the Al
137+ Keggin complex in the liquid state, it was only possible to compare the relevant characteristics of this complex with those of the investigated in the present work Al
132+ in the crystal state. The standard enthalpy change of formation from elements of the Al
132+ complex in the crystal state is equal to −10,445.6 ± 10.9 kJ/mol, which is almost twice as low as the standard enthalpy change of formation from elements of the Al
137+ Keggin complex in the crystal state (−20892.39 ± 70.56 kJ/mol), determined by Armstrong et al. [
43]. This observation indicates the higher reactivity/lower stability of the Al
132+ complex in comparison to Al
137+ Keggin complex, and proves the hypothesis that the latter complex in its usual form is less likely to participate in any reactions [
46].
The results acquired during the testing of different limit noises for the applicability of the derived PLSR model indicate that this model can be used, not merely for the computational results, but also for the experimentally-obtained data with the maximum noise, which affects the instrument amplification, 6%, and the maximum standard deviation for the noise, which affects the measurement signal, equal to 14, imposed on all the investigated variables (ΔHo, ΔSo, ΔGo).
The performed PLSR and cluster analyses are the bases for developing a classifier that enables a continuous discrimination of the thermodynamically stable and less stable Al13-monosaccharide complexes, based on the coagulant type and the content of the monosaccharide species in mixed liquor in the separation chamber of the MBR/BF-MBR system, which will considerably contribute to plant surveillance and process control.
The concept, presented in the current study, provides a physicochemical and statistical verification of the reasons for the observations on the SMPs removal, which were obtained during the filtration tests in BF-MBR, conducted earlier [
31].
The developed approach will help to reduce the input (time and resources), required to test the coagulants as the fouling reducers in MBR and BF-MBR systems. It addresses the questions “What to expect from the mixed liquor system with the chemical dosing?” and “How to reduce the mixed liquor fouling potential?” The quantum chemical and thermodynamic calculations, followed by the multivariate chemometric analysis, allow the researcher to predict the efficiency of the coagulants with regard to SMPs removal in MBR and BF-MBR, and to select the most efficient compound without conducting numerous experiments. The thermodynamic stability of the investigated Al
13-monosaccharide complexes is directly related to their fouling potential, since if the complex is more liable to decompose during/after coagulation, more severe membrane fouling is expected due to the released SMPs [
97]. Hence, the present study introduces the relationship—fouling as a function of the standard Gibbs energy change—which is able to provide the best solution for the selection of the chemical fouling reducer in MBR and BF-MBR at the lowest cost.
The results of the present work can be used for the development of a sensor for the prediction of the flux enhancement efficiency based on the monosaccharide content of mixed liquor in BF-MBR.
Further study is also foreseen in the investigation of the role of the stabilizing factors in the formation of Al13-monosaccharide complexes.
Besides, the thermodynamic properties of the interaction of the monosaccharide-amino acid network assemblages with the polyaluminum hydrolysis species should be investigated.
4. Conclusions
The developed computational thermodynamic-multivariate chemometric approach to the assessment of the chemical flux enhancement of the coagulants with regard to MBR and BF-MBR advances the field of fouling mitigation, and provides the thermodynamic and statistical tools for the understanding of the underlying mechanisms and the prediction of the fouling mitigation efficiency.
β-D-glucopyranose, β-D-galactopyranose, β-D-mannopyranose, α-D-glucopyranuronic acid, β-L-rhamnopyranose, β-L-fucopyranose, α-L-arabinofuranose, and β-D-glucosamine are the main building blocks of SMPs of mixed liquor in the MBR/BF-MBR, and are primarily responsible the reversible, irreversible and irrecoverable membrane fouling.
The case study, presented in the current work, focuses on the flux enhancement efficiency of the Al132+ complex, which is the main hydrolysis species of the medium basicity prepolymerized aluminum chloride, proven to be the most efficient coagulant with respect to fouling mitigation in BF-MBR.
The mechanisms of the formation of the most thermodynamically stable Al13-monosaccharide complexes were defined based on the results of the quantum chemical and thermodynamic analyses. The thermodynamic stability of the formed Al13-monosaccharide complex was found to be highly dependent upon the active centers of the monosaccharides and the Al132+ complex, which participate in the chemical coordination process, and the nature of the monosaccharide. The complex, formed by the interaction of the oxygen atoms at C4−C6 atoms of α-D-glucopyranuronic acid with the Al atoms no. 69, 73 of the Al132+ complex, was identified as the most thermodynamically stable complex (ΔGo = −1398.87 kJ/mol). Meanwhile, the complex, formed by the interaction of the oxygen atoms at C1C2 atoms of β-L-rhamnopyranose with the Al atoms no. 23, 10 of the Al132+ complex, exhibited the lowest thermodynamic stability among the investigated monosaccharides (ΔGo = −1067.79 kJ/mol).
The sample grouping. according to the standard Gibbs energy change, performed during the PLSR analysis, revealed that the complexes formed by α-D-glucopyranuronic acid and the Al atoms no. 69, 73; 62, 37; and 69, 62 of the Al132+ complex make up the majority of the most thermodynamically stable samples among all 256 investigated Al13-monosaccharide complexes.
The derived PLSR model can be used for computationally and experimentally acquired thermodynamic parameters, being applicable (R-squared = 0.7) for the maximum proportional noise equal to 6%, and the maximum standard deviation for the additive noise equal to 14, applied for both the predictors (ΔHo, ΔSo) and the response function (ΔGo).
The results of the performed PLSR and cluster analyses are the basis for developing a classification model for the continuous discrimination of the thermodynamically stable and less stable Al13-monosaccharide complexes, based on the coagulant type and content of the monosaccharide species in mixed liquor, which will substantially contribute to the automated process control of the MBR/BF-MBR systems.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}