Stabilization of Chromium (VI) in the Presence of Iron (II): Method Development and Validation

Abstract
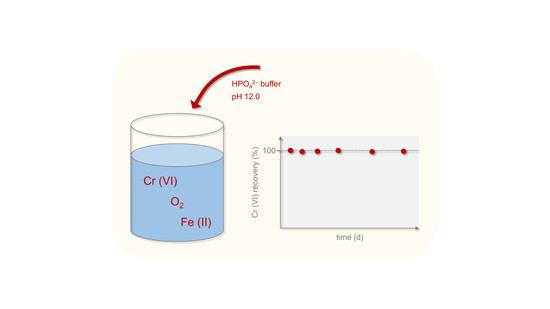
1. Introduction
2. Materials and Methods
2.1. Chemicals and Stock Solutions
2.2. Stabilization by Inorganic Buffer Systems
2.3. Stabilization by Chelating Agents
2.4. Analyses
2.5. Correlation Analysis
3. Results and Discussion
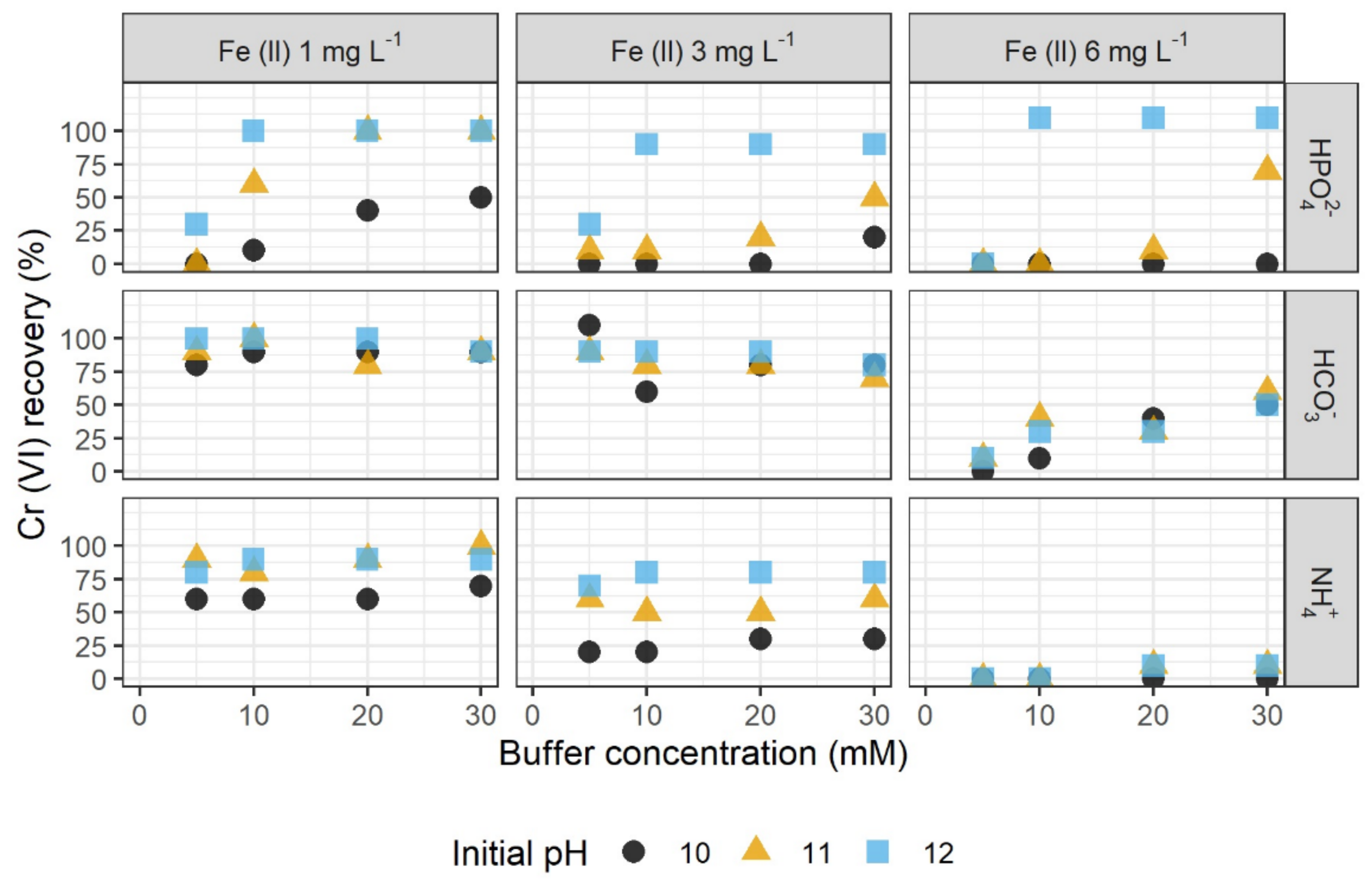
3.1. Stabilization by Inorganic Buffer Systems
3.1.1. Influencing Parameters
3.1.2. XRD Analysis of Precipitation Products
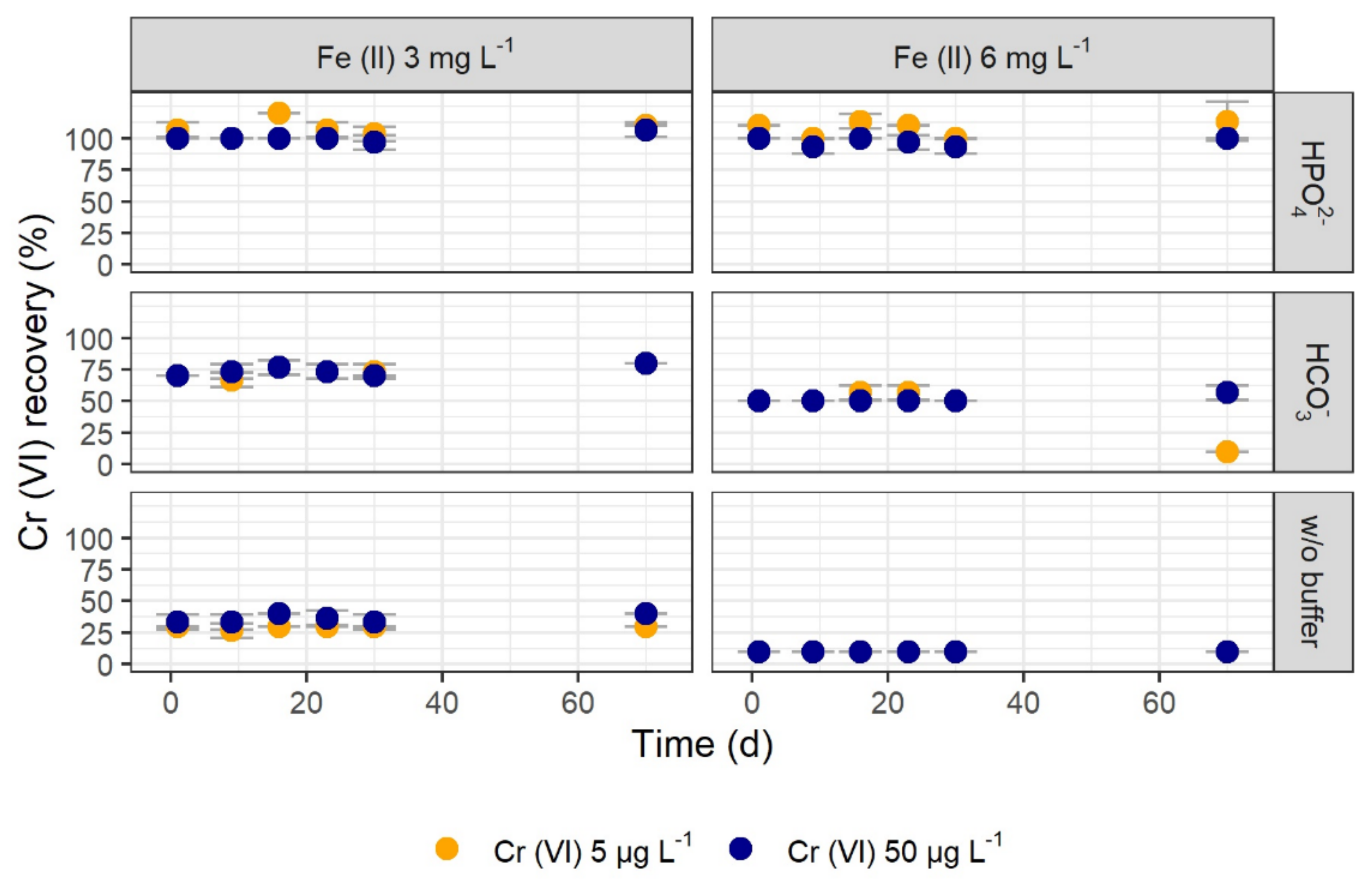
3.1.3. Long-Term Tests
3.2. Stabilization by Chelating Agents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnhart, J. Occurrences, uses, and properties of chromium. Regul. Toxicol. Pharmacol. 1997, 26, S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Schewel, L.; Graedel, T.E. The contemporary anthropogenic chromium cycle. Environ. Sci. Technol. 2006, 40, 7060–7069. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, J.J.; Bansal, N.; Chirwa, E.M.N. Chromium in environment, its toxic effect from chromite-mining and ferrochrome industries, and its possible bioremediation. Expo. Health 2018, 12, 51–62. [Google Scholar] [CrossRef]
- Costa, M.; Klein, C.B. Toxicity and carcinogenicity of chromium compounds in humans. Crit. Rev. Toxicol. 2006, 36, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.J.. Chromium cycling in soils and water: links, gaps, and methods. Environ. Health Perspect. 1991, 92, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Loyaux-Lawniczak, S.; Lecomte, P.; Ehrhardt, J.-J. Behavior of hexavalent chromium in a polluted groundwater: redox processes and immobilization in soils. Environ. Sci. Technol. 2001, 35, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Petrusevski, B.; Amy, G. Chromium removal from water: A review. J. Water Supply Res. Technol.-Aqua 2008, 57, 541–553. [Google Scholar] [CrossRef]
- Anderson, R.A. Chromium as an essential nutrient for humans. Regul. Toxicol. Pharmacol. 1997, 26, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Costa, M. Toxicity and carcinogenicity of Cr(VI) in animal models and humans. Crit. Rev. Toxicol. 1997, 27, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Roller, M. Potentielle Schädlichkeit von Chrom im Trinkwasser—Bericht zum Sondervorhaben des Umweltbundesamtes. FKZ 363 01 399, 2012. [Google Scholar]
- Council of the European Union. Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption. Available online: https://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX%3A31998L0083 (accessed on 24 March 2020).
- US EPA 40 CFR 141—National Primary Drinking Water Regulations. Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 28 February 2020).
- Federal Department of Home Affairs, Switzerland. Verordnung des EDI vom 16. Dezember 2016 über Trinkwasser sowie Wasser in öffentlich zugänglichen Bädern und Duschanlagen (TBDV). Available online: https://www.admin.ch/opc/de/classified-compilation/20143396/index.html (accessed on 24 March 2020).
- World Health Organization (WHO). Guidelines for Drinking-water Quality; 2017; ISBN 978-92-4-154995-0. Available online: https://www.who.int/water_sanitation_health/publications/drinking-water-quality-guidelines-4-including-1st-addendum/en/ (accessed on 24 March 2020).
- Rivero-Huguet, M.; Marshall, W.D. Influence of various organic molecules on the reduction of hexavalent chromium mediated by zero-valent iron. Chemosphere 2009, 76, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Melitas, N.; Chuffe-Moscoso, O.; Farrell, J. Kinetics of soluble chromium removal from contaminated water by zerovalent iron media: corrosion inhibition and passive oxide effects. Environ. Sci. Technol. 2001, 35, 3948–3953. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Hering, J.G. Removal of chromium(VI) from drinking water by redox-assisted coagulation with iron(II). J. Water Supply Res. Technol.-Aqua 2003, 52, 319–332. [Google Scholar] [CrossRef]
- Eary, L.E.; Rai, D. Chromate removal from aqueous wastes by reduction with ferrous ion. Environ. Sci. Technol. 1988, 22, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Pettine, M.; D’Ottone, L.; Campanella, L.; Millero, F.J.; Passino, R. The reduction of chromium (VI) by iron (II) in aqueous solutions. Geochim. Cosmochim. Acta 1998, 62, 1509–1519. [Google Scholar] [CrossRef]
- Schlautman, M.A.; Han, I. Effects of pH and dissolved oxygen on the reduction of hexavalent chromium by dissolved ferrous iron in poorly buffered aqueous systems. Water Res. 2001, 35, 1534–1546. [Google Scholar] [CrossRef]
- Fendorf, S.E.; Li, G. Kinetics of chromate reduction by ferrous iron. Environ. Sci. Technol. 1996, 30, 1614–1617. [Google Scholar] [CrossRef]
- Buerge, I.J.; Hug, S.J. Kinetics and pH dependence of chromium(VI) reduction by iron(II). Environ. Sci. Technol. 1997, 31, 1426–1432. [Google Scholar] [CrossRef]
- Christmann, E.P.; Badgett, J.L. Interpreting Assessment Data: Statistical Techniques You Can Use; NSTA Press: Arlington, VA, USA, 2009; ISBN 978-1-933531-36-6. [Google Scholar]
- Federal Ministry of Health (Germany). German Ordinance on the Quality of Water intended for Human Consumption. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=3&cad=rja&uact=8&ved=2ahUKEwj1gpqh77PoAhUCzKQKHSJmDaIQFjACegQIBRAB&url=https%3A%2F%2Fwww.bundesgesundheitsministerium.de%2Ffileadmin%2FDateien%2F3_Downloads%2FE%2FEnglische_Dateien%2FDrinking_Water_Ordinance.pdf&usg=AOvVaw0-5xASQNS_p00R09JELUNd (accessed on 24 March 2020).
- Tamura, H.; Goto, K.; Nagayama, M. Effect of anions on the oxygenation of ferrous ion in neutral solutions. J. Inorg. Nucl. Chem. 1976, 38, 113–117. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 87th ed.; Taylor & Francis: Boca Raton, FL, USA, 2006; ISBN 978-0-8493-0487-3. [Google Scholar]
- Kalka, H. Solubility product constants Ksp at 25 °C. Available online: https://www.aqion.de/site/16 (accessed on 18 March 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer System | Fe (II) (mg L– 1) | Water | Identified Compounds |
|---|---|---|---|
| HPO42– | 100 | process | amorphous peak, iron hydroxide |
| HCO3– | 100 | pure | trona, nahcolite |
| 50 | pure | trona, nahcolite | |
| 100 | process | calcite, nahcolite | |
| 6 | process | calcite, trona, nahcolite, quartz |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahringer, D.; Polenz, C.; El-Athman, F. Stabilization of Chromium (VI) in the Presence of Iron (II): Method Development and Validation. Water 2020, 12, 924. https://doi.org/10.3390/w12040924
Mahringer D, Polenz C, El-Athman F. Stabilization of Chromium (VI) in the Presence of Iron (II): Method Development and Validation. Water. 2020; 12(4):924. https://doi.org/10.3390/w12040924
Chicago/Turabian StyleMahringer, Daniel, Chantal Polenz, and Fatima El-Athman. 2020. "Stabilization of Chromium (VI) in the Presence of Iron (II): Method Development and Validation" Water 12, no. 4: 924. https://doi.org/10.3390/w12040924
APA StyleMahringer, D., Polenz, C., & El-Athman, F. (2020). Stabilization of Chromium (VI) in the Presence of Iron (II): Method Development and Validation. Water, 12(4), 924. https://doi.org/10.3390/w12040924