2.2. Slag Source, Preparation, Characterization
The slag material was from an electric-arc furnace (EAF) steel mill in Butler, Indiana, USA. Two different EAF slag sizes were obtained, the fine (1 to 5 mm) and coarse (10 to <50 mm) fractions. The coarse size fraction was considered inert and used for the purpose of physical drainage, not chemical P removal. For the remainder of this paper, unless specified otherwise, all references to slag in the blind inlet and for laboratory analysis is assumed to be of the “fine” 1–5 mm fraction. Slag was sieved at the steel plant by the slag-handling company (Edwin Levy Co., Dearborn, MI, USA). After delivery to the site, a subsample of EAF slag was taken for physical and chemical characterization. Briefly, particle size distribution was determined via sieve analysis and bulk density was measured by weighing a known volume of slag. Total elemental analysis of the EAF slag was achieved using heated acid digestion and followed by analysis of the digestate by inductively coupled plasma atomic emission spectroscopy (ICP-AES) for Ca, Mg, Fe, Al, Mn, K, Cr, Ni, Pb, Zn, Cu, and S [
12]. pH and electrical conductivity (EC) were measured in de-ionized (DI) water with a pH and EC meter after equilibrating slag for 30 min in a 1:5 solid/solution.
Slag was tested for P removal under lab conditions using a flow-through cell [
7,
9]. Briefly, two tests were conducted in duplicate, in which 0.2 and 2.5 mg P L
−1 was supplied via a Mariotte bottle to a flow-through cell containing 2 g of slag. The flow rate was adjusted to achieve a 10-min retention time (retention time = pore volume divided by flow rate) using a peristaltic pump located downstream/below the cell. Outflow samples were collected every 2 h and analyzed for dissolved P using the colorimetric method described in a later section. Inflow concentrations of 2.5 and 0.2 mg L
−1 were chosen to represent the magnitude of P concentration from runoff when poultry litter was recently applied, and also after soil equilibration, as observed in field data.
2.5. Chemical Analysis of Water Samples
Water samples were vacuum-filtered through 0.45-μm nylon filters with a 20-mL aliquot acidified with 100 μL of 50% H
2SO
4 solution. These samples were used to determine dissolved P, NH
4-N, and NO
3-N colorimetrically using a Thermo Scientific Gallery autoanalyzer (Thermo Scientific, Waltham, MA, USA) following USEPA methods 365.2, 350.1 rev 2, and 353.1, respectively [
14]; the limits of detection were 0.005, 0.01, and 0.01 mg L
−1, respectively.
Total Kjeldahl nitrogen (TKN) and total phosphorus (TP) were determined in unfiltered, acidified (60 mL sample + 100 μL of 50% H
2SO
4 solution) water samples following EPA methods 351.2 rev. 2 and 365.4, respectively [
14]. Briefly, samples were digested with mercuric sulfate on a Lachat BD-46 block digestor (Hach, Loveland, CO, USA). Then, TKN and TP were determined in a Thermo Fisher Konelab colorimetric autoanalyzer (Thermo Scientific, Waltham, MA, USA). The limits of detection for both TKN and TP were 0.01 mg L
−1.
Dissolved C content was determined in filtered (0.45-μm nylon filters) and non-acidified samples using wet chemical oxidation and non-dispersive infrared detection of CO2 with a Shimadzu TOC-Vws analyzer (Shimadzu Inc., Kyoto, Japan). Total dissolved C (TDC) in the sample was determined by oxidizing all C with sodium persulfate and phosphoric acid to produce CO2. Inorganic C (IC) content was determined by adding phosphoric acid to the sample to produce CO2. Potassium hydrogen phthalate and sodium bicarbonate were used as external standards (range from 5 to 80 mg L−1) for TDC and IC, respectively. The dissolved organic C (DOC) content was calculated as the difference between TDC and IC.
Soluble K+, Mg2+, Ca2+, and SO42−-S were determined in unacidified syringe-filtered (0.45-μm nylon filter) water samples using two 2100 Dionex ion chromatography systems (Thermo Scientific, Waltham, MA, USA) equipped with eluent generators and electrical conductivity detectors. An ion chromatography system was dedicated to the analysis of cations and the other for anions, where these two systems shared an AS-AP ICS-5000 autosampler (50-μL sample injection for each). A 250 mm length × 4 mm diameter Dionex IonPac CS12A ion column was used to separate the cations with isocratic 20 mM methanesulfonic acid as eluent with a flow rate of 1 mL min−1. After the separation of cations, the eluent current was suppressed with a CSRS 300 4-mm suppressor (Thermo Scientific, Waltham, MA, USA) set at 59 mA. Then, the electrical current of each cation was measured with a DS6 Dionex electrical conductivity cell set at 30 °C. Identification and quantification of cations were performed using a mixture of external standards. Calibration curves were performed with concentrations ranging from 0.06 to 40 mg L−1 and using the 1/concentration weighting linear curve fitting. Retention times of K+, Mg2+, and Ca2+ were 6.73, 10.37, and 13.51 min, respectively. Separation of anions was performed with a 250 mm length × 4 mm diameter Dionex IonPac AS18 column set at 40 °C and a KOH solution gradient as eluent (10 mM hold from the 1 min, ramp to 35 mM for 8 min, ramp to 45 mM for 1 min and hold for 5 min, then 10 mM for 3 min). The flow rate of the eluent was 1.2 mL min−1. An ASRS-ULTRA II 4-mm suppressor (Thermo Scientific, Waltham, MA, USA), set at 134 mA, was used to suppress the electrical current of the eluent in the anion analysis. The electrical conductivity cell was set at 40 °C to measure the electrical current of the anions. Like the cations, external standards (0.05 to 40 mg L−1) were used to perform calibration curves with 1/concentration weighting linear curve fitting and to quantify the amount of anions. Retention time of SO42−-S was 8.79 min.
The herbicide atrazine and its metabolites deethylatrazine (DEA), deisopropylatrazine (DIP), and 2-hydroxyatrazine (OH-ATZ) were analyzed in unacidified syringe-filtered (0.45-μm nylon filter) water samples using a Waters Acquity ultra-performance liquid chromatography (UPLC) system (Milford, MA, USA) coupled with a Waters Acquity TQ tandem quadrupole mass (MS) detector. The separation of the compounds was performed with a 100 mm × 2.1 mm × 1.7 μm Waters Acquity UPLC BEH C18 column using a mobile phase gradient with 0.01% formic acid (A) and acetonitrile (B) and flow rate of 0.45 mL min−1. The initial gradient was: 30% B, hold for 0.70 min, increase to 60% B in 4.8 min, increase to 75% B in 0.5 min, hold for 0.5 min, decrease to 30% B in 0.5 min, and hold for 0.5 min. All solvents were Optima grade (Thermo Fisher Scientific Inc., Waltham, MA, USA). The MS detector was set in positive ionization, and the Multiple Reaction Monitoring mode was used for the detection and confirmation of compounds. The MS voltages in the capillary, cone, extractor, and radio frequency lens were 0.61, 40, 3.0, and 0.1 kV, respectively. Source and desolvation temperatures were set at 150 and 400 °C, respectively. Desolvation and cone gas flows were set at 850 and 20 L h−1, respectively. For each compound, the mass-to-charge ratio (m/z) of the parent material was used for quantification, and the most prominent fragment was used for confirmation of the compound: Atrazine: 216 > 156 > 174; DEA: 188 > 146 > 104; DIP: 174 > 104 > 132; OH-ATZ: 198 > 156 > 86. External standards (0.05 to 40 μg L−1) were prepared in 25% methanol. The calibration curve was forced to the origin, fitted to the quadratic form, and 1/concentration as the weighted function. To minimize introduction of salts from the samples to the MS detector, samples were diluted as follows: 0.5 mL of sample + 0.45 mL of nanopure water + 0.05 mL of 100 μg L−1 D5-atrazine (as internal standard) were added to a 1.5-mL vial and vortexed before analysis. The internal standard (m/z quantification and confirmation 221 > 101 > 69) was also added to the external standards. The retention times of atrazine, D5-atrazine, DEA, DIP, and OH-ATZ were 2.90, 2.90, 1.20, 0.93, and 0.77 min, respectively.
Dicamba and glyphosate (and its metabolite aminomethyl phosphonic acid, AMPA) were analyzed by ion chromatography as anions using the same method for SO42−-S; the retention times were 15.33, 12.70, and 7.05 min, respectively. The ranges of the calibration curves were from 0.01 to 5.00 mg L−1 for dicamba, from 0.025 to 5.00 mg L−1 for glyphosate, and 0.10 to 5.00 mg L−1 for AMPA.


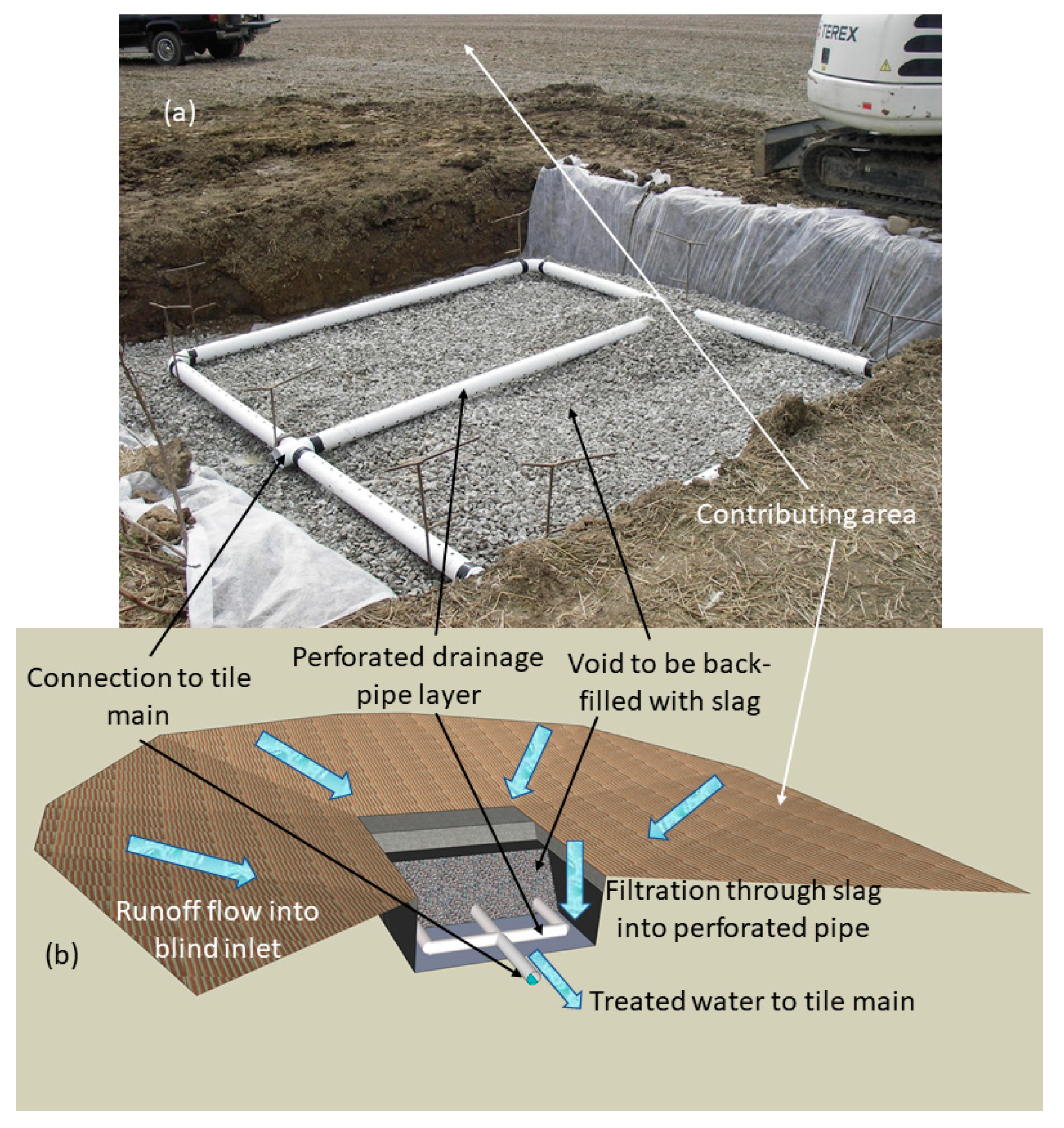



{kind=link}
{kind=link}
{kind=link}