
Soil Microbiome Study Based on DNA Extraction: A Review


Abstract
1. Introduction
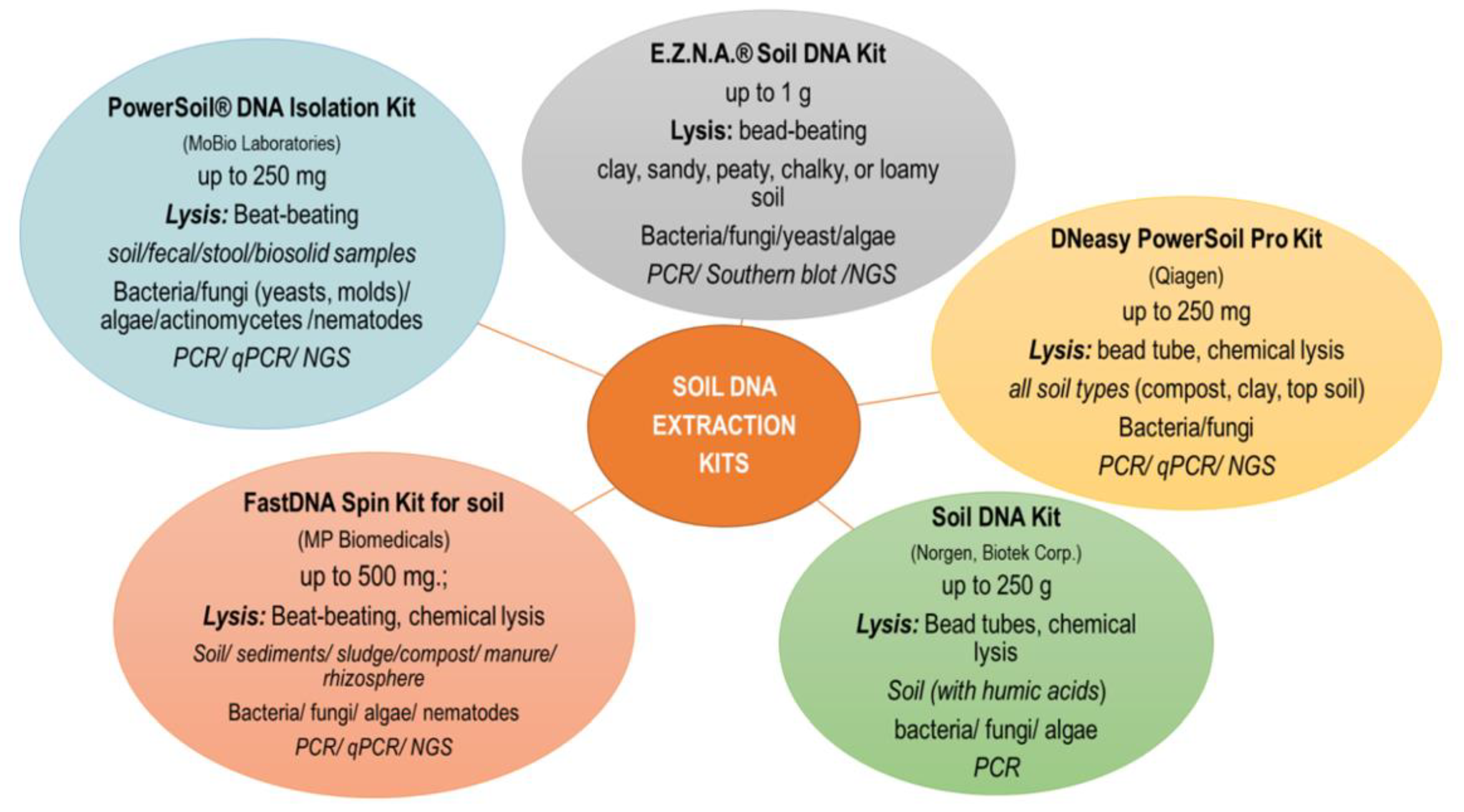
2. Extraction DNA
2.1. Methods of DNA Extraction
2.2. The Main Factors Influencing on Yield of DNA Extraction and Its Downstream of Application
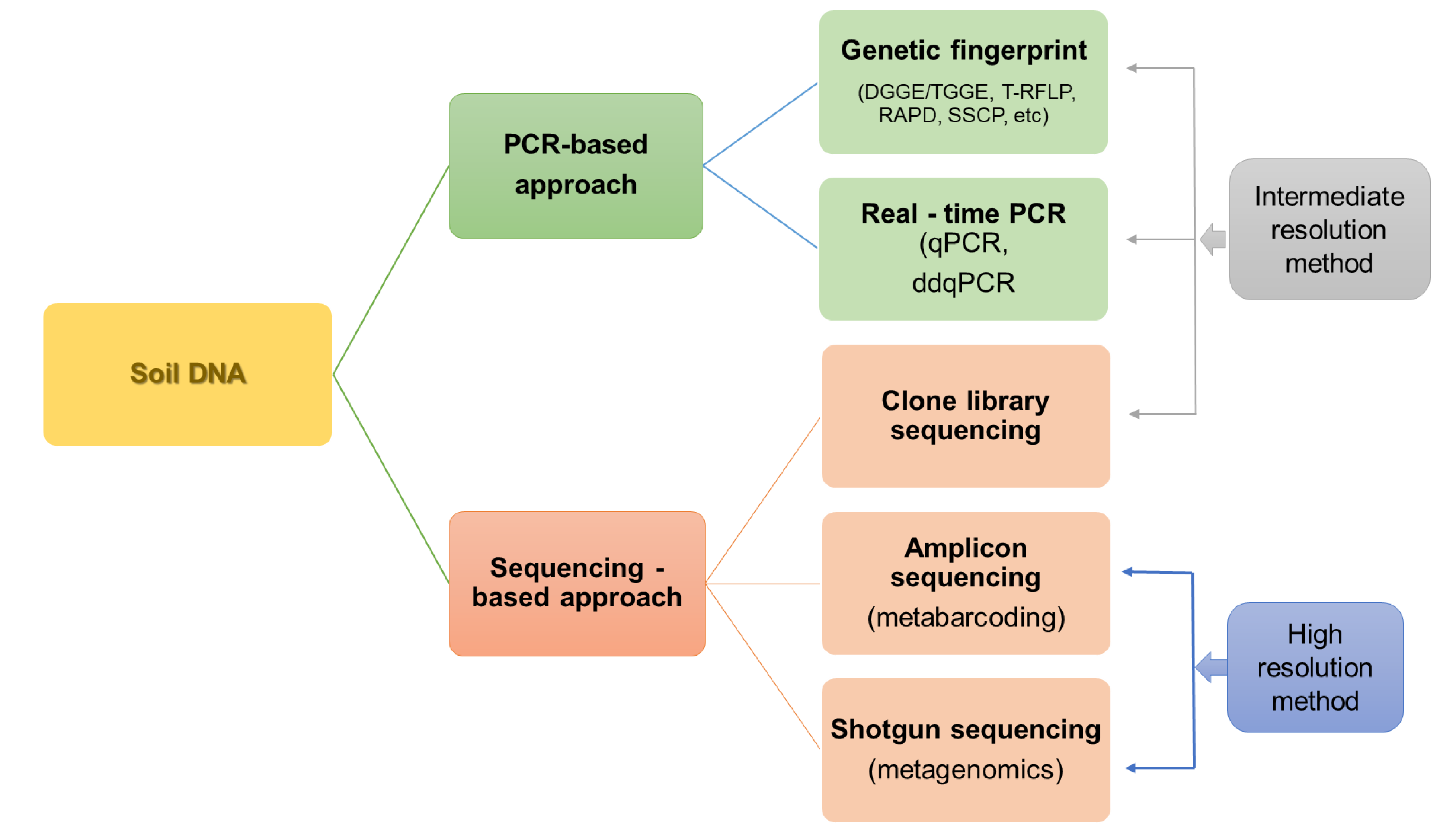
3. Soil DNA-Based Methods
3.1. Partial Analysis of Soil Microbiota
3.2. High-Throughput Sequencing Techniques
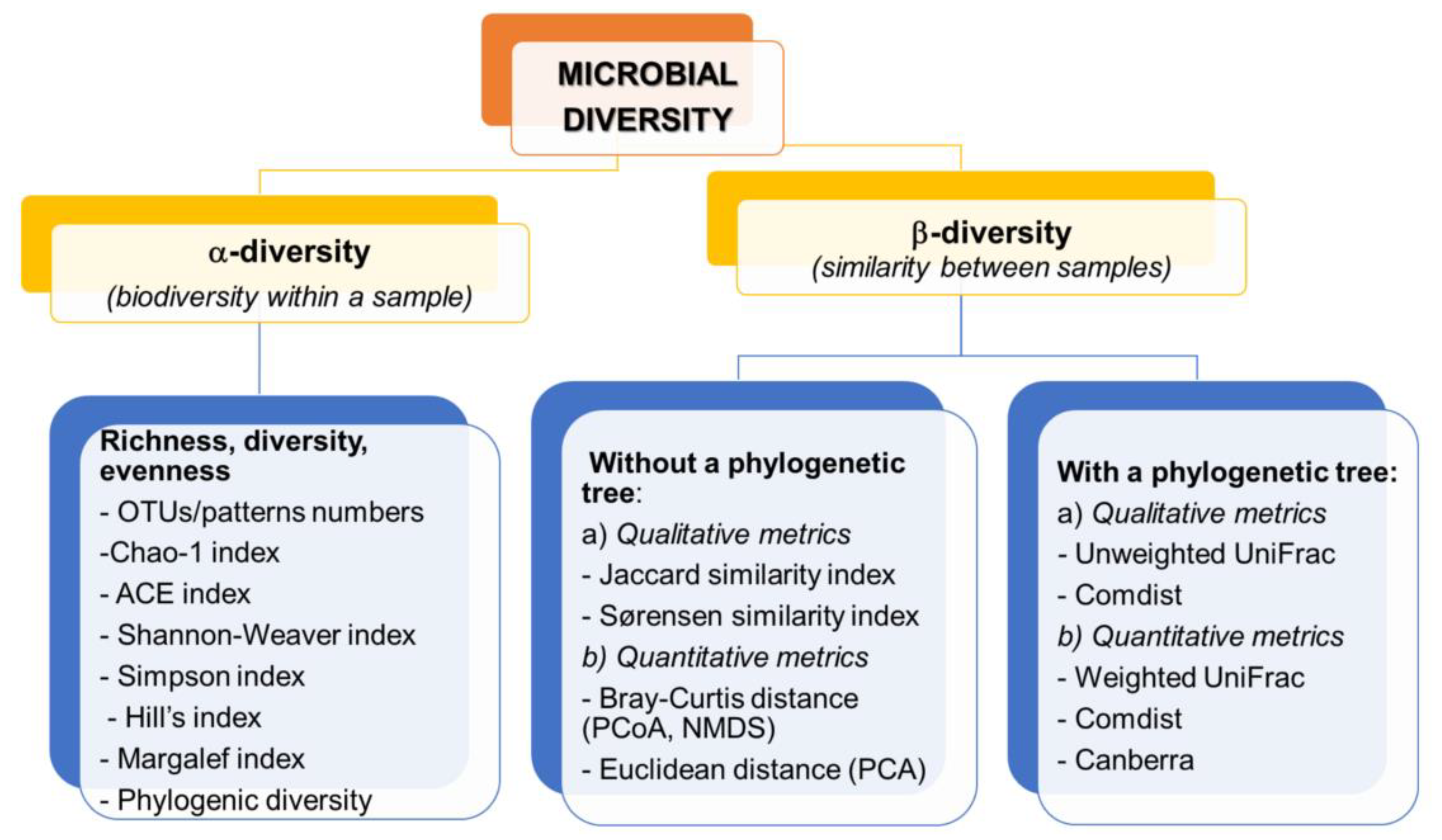
4. Bioinformatics and Statistical Analysis in Diversity Estimation
4.1. Bioinformatics
4.2. Statistics
5. Genetic Diversity of Soil Microorganisms—Application Areas
5.1. Fertilization Strategies, Agricultural Soil Monitoring
5.2. Soil Contamination and Remediation Monitoring
5.3. Effect of Pesticides and Another Organic Xenobiotis
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cornea, C.P.; Voaideş, C.; Ciuca, M.; Stan, V.; Gament, E.; Razec, I.; Duşa, M. Molecular Methods for Assessement the Bacterial Communities from Different Type of Soils in Romania. Not. Bot. Horti Agrobot. Cluj-Napoca 2011, 39, 64–70. [Google Scholar] [CrossRef]
- Wołejko, E.; Jabłońska-Trypuć, A.; Wydro, U.; Butarewic, Z.A.; Łozowicka, B. Soil biological activity as an indicator of soil pollution with pesticides—A review. Appl. Soil. Ecol. 2020, 147, 103356. [Google Scholar] [CrossRef]
- Singh, B.K.; Campbell, C.D.; Sorenson, S.J.; Zhou, J. Soil genomics. Nat. Rev. Microbiol. 2009, 7, 756. [Google Scholar] [CrossRef] [PubMed]
- Kaden, R.; Krolla-Sidenstein, P. How to Show the Real Microbial Biodiversity? A Comparison of Seven DNA Extraction Methods for Bacterial Population Analyses in Matrices Containing Highly Charged Natural Nanoparticles. Microorganisms 2015, 3, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Smalla, K.; Oros-Sichler, M.; Milling, A.; Heuer, H.; Baumgarte, S.; Becker, R.; Neuber, G.; Kropf, S.; Ulrich, A.; Tebbe, C.C. Bacterial diversity of soils assessed by DGGE, T-RFLP and SSCP fingerprints of PCR-amplified 16S rRNA gene fragments: Do the different methods provide similar results? J. Microbiol. Methods 2007, 69, 470–479. [Google Scholar] [CrossRef]
- Tanveer, A.; Yadav, S.; Yadav, D. Comparative assessment of methods for metagenomic DNA isolation from soils of different crop growing fields. 3 Biotech 2016, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Singh, H.; Sharma, P.C. An improved method suitable for isolation of high-quality metagenomic DNA from diverse soils. 3 Biotech 2017, 7, 171. [Google Scholar] [CrossRef]
- Salonen, A.; Nikkilä, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilić-Stojanović, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative Analysis of Fecal DNA Extraction Methods with Phylogenetic Microarray: Effective Recovery of Bacterial and Archaeal DNA Using Mechanical Cell Lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
- Delmont, T.O.; Robe, P.; Clark, I.; Simonet, P.; Vogel, T.M. Metagenomic Comparison of Direct and Indirect Soil DNA Extraction Approaches. J. Microbiol. Methods 2011, 86, 397–400. [Google Scholar] [CrossRef]
- Mazziotti, M.; Henry, S.; Laval-Gilly, P.; Bonnefoy, A.; Falla, J. Comparison of Two Bacterial DNA Extraction Methods from Non-Polluted and Polluted Soils. Folia Microbiol. 2018, 63, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, G.; Sani, R.K. Molecular Techniques to Assess Microbial Community Structure, Function, and Dynamics in the Environment. In Microbes and Microbial Technology; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer: New York, NY, USA, 2011; pp. 29–57. [Google Scholar] [CrossRef]
- Abed, R.M.; Safi, N.M.D.; Köster, J.; de Beer, D.; El-Nahhal, Y.; Rullkötter, Y.; Garcia-Pichel, F. Microbial Diversity of a Heavily Polluted Microbial Mat and Its Community Changes following Degradation of Petroleum Compounds. Appl. Environ. Microbiol. 2002, 68, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- George, P.B.L.; Lallias, D.; Creer, S.; Seaton, F.M.; Kenny, J.G.; Eccles, R.M.; Griffiths, R.I.; Lebron, I.; Emmett, B.A.; Robinson, D.A.; et al. Divergent national-scale trends of microbial and animal biodiversity revealed across diverse temperate soil ecosystems. Nat. Commun. 2019, 10, 1107. [Google Scholar] [CrossRef] [PubMed]
- Sidstedt, M.; Rådström, P.; Hedman, J. PCR inhibition in qPCR, dPCR and MPS—mechanisms and solutions. Anal. Bioanal. Chem. 2020, 412, 2009–2023. [Google Scholar] [CrossRef] [PubMed]
- Semenov, M.V. Metabarcoding and Metagenomics in Soil Ecology Research: Achievements, Challenges, and Prospects. Biol. Bull. Rev. 2021, 11, 40–53. [Google Scholar] [CrossRef]
- Liu, W.; Liu, L.; Yang, X.; Deng, M.; Wang, Z.; Wang, P.; Yang, S.; Li, P.; Peng, Z.; Yang, L.; et al. Long-Term Nitrogen Input Alters Plant and Soil Bacterial, but Not Fungal Beta Diversity in a Semiarid Grassland. Glob. Chang. Biol. 2021, 27, 3939–3950. [Google Scholar] [CrossRef]
- Socolar, J.B.; Gilroy, J.J.; Kunin, W.E.; Edwards, D.P. How Should Beta-Diversity Inform Biodiversity Conservation? Trends Ecol. Evol. 2016, 31, 67–80. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular Ecological Network Analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Ren, C.; Zhang, W.; Zhong, Z.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Differential responses of soil microbial biomass, diversity, and compositions to altitudinal gradients depend on plant and soil characteristics. Sci. Total Environ. 2018, 610–611, 750–758. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Souza, M.D.F.; de Laia, M.L.; Melo, J.D.O.; da Costa, M.R.; Gonçalves, J.F.; Silva, D.V.; dos Santos, J.B. Metagenomic analysis reveals mechanisms of atrazine biodegradation promoted by tree species. Environ. Pollut. 2020, 267, 115636. [Google Scholar] [CrossRef]
- García-Lozano, M.; Hernández-De Lira, I.O.; Huber, D.H.; Balagurusamy, N. Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes 2019, 7, 408. [Google Scholar] [CrossRef]
- Du, Z.; Zhu, Y.; Zhu, L.; Zhang, J.; Li, B.; Wang, J.; Wang, J.; Zhang, C.; Cheng, C. Effects of the herbicide mesotrione on soil enzyme activity and microbial communities. Ecotoxicol. Environ. Saf. 2018, 164, 571–578. [Google Scholar] [CrossRef]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 2020, 61, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Genet. 2018, 16, 410–422. [Google Scholar] [CrossRef]
- Ghosh, A.; Bhadury, P. Methods of Assessment of Microbial Diversity in Natural Environments. In Microbial Diversity in the Genomic Era, 1st ed.; Das, S., Dash, H., Eds.; Academic Press, Elsevier: London, UK, 2019; pp. 3–14. [Google Scholar] [CrossRef]
- Kumar, R.; Joshi, S.R. Microbial Ecology of Soil: Studying the diversity of microorgansims in the most complex of the environments. A review. Adv. Appl. Microbiol. 2015, 19, 267–279. [Google Scholar]
- Nannipieri, P.; Penton, C.R.; Purahong, W.; Schloter, M.; van Elsas, J.D. Recommendations for soil microbiome analyses. Biol. Fertil. Soils 2019, 55, 765–766. [Google Scholar] [CrossRef]
- Dubey, R.K.; Tripathi, V.; Prabha, R.; Chaurasia, R.; Singh, D.P.; Rao, C.S.; El-Keblawy, A.; Abhilash, P.C. Bioinformatics Tools for Soil Microbiome Analysis. In Unravelling the Soil Microbiome, 1st ed.; SpringerBriefs in Environmental Science; Springer: Cham, Switzerland, 2020; pp. 61–70. [Google Scholar] [CrossRef]
- Niu, S.-Y.; Yang, J.; McDermaid, A.; Zhao, J.; Kang, Y.; Ma, Q. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Briefings Bioinform. 2017, 19, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Sadowsky, M.J. Practical considerations for sampling and data analy-sis in contemporary metagenomics-based environmental studies. J. Microbiol. Methods 2018, 154, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA Recovery from Soils of Diverse Composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y. Improvements in Extraction Methods of High-molecular-weight DNA from Soils by Modifying Cell Lysis Conditions and Reducing Adsorption of DNA onto Soil Particles. Microbes Environ. 2021, 36, ME21017. [Google Scholar] [CrossRef]
- Philippot, L.; Abbate, C.; Bispo, A.; Chesnot, T.; Hallin, S.; Lemanceau, P.; Lindström, K.; Pandard, P.; Romero, E.; Schloter, M.; et al. Soil microbial diversity: An ISO standard for soil DNA extraction. J. Soils Sediments 2010, 10, 1344–1345. [Google Scholar] [CrossRef]
- Young, J.M.; Rawlence, N.J.; Weyrich, L.S.; Cooper, A. Limitations and Recommendations for Successful DNA Extraction from Forensic Soil Samples: A Review. Sci. Justice 2014, 54, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Fatima, F.; Pathak, N.; Rastogi Verma, S. An Improved Method for Soil DNA Extraction to Study the Microbial Assortment within Rhizospheric Region. Mol. Biol. Int. 2014, 2014, 518960. [Google Scholar] [CrossRef] [PubMed]
- Knauth, S.; Schmidt, H.; Tippkötter, R. Comparison of Commercial Kits for the Extraction of DNA from Paddy Soils. Lett. Appl. Microbiol. 2013, 56, 222–228. [Google Scholar] [CrossRef]
- Felczykowska, A.; Krajewska, A.; Zielińska, S.; Łoś, J.M. Sampling, Metadata and DNA Extraction—Important Steps in Metagenomic Studies. Acta Biochim. Pol. 2015, 62, 151–160. [Google Scholar] [CrossRef]
- Kuske, C.R.; Banton, K.L.; Adorada, D.L.; Stark, P.C.; Hill, K.K.; Jackson, P.J. Small-Scale DNA Sample Preparation Method for Field PCR Detection of Microbial Cells and Spores in Soil. Appl. Environ. Microbiol. 1998, 64, 2463–2472. [Google Scholar] [CrossRef]
- Krsek, M.; Wellington, E.M. Comparison of Different Methods for the Isolation and Purification of Total Community DNA from Soil. J. Microbiol. Methods 1999, 39, 1–16. [Google Scholar] [CrossRef]
- Islam, M.R.; Sultana, T.; Melvin Joe, M.; Cho, J.-C.; Sa, T. Comparisons of Direct Extraction Methods of Microbial DNA from Different Paddy Soils. Saudi J. Biol. Sci. 2012, 19, 337–342. [Google Scholar] [CrossRef][Green Version]
- Gaur, M.; Vasudeva, A.; Singh, A.; Sharma, V.; Khurana, H.; Negi, R.K.; Lee, J.-K.; Kalia, V.C.; Misra, R.; Singh, Y. Comparison of DNA Extraction Methods for Optimal Recovery of Metagenomic DNA from Human and Environmental Samples. Indian J. Microbiol. 2019, 59, 482–489. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Olson, B.H. Rapid Method for Direct Extraction of DNA from Soil and Sediments. Appl. Environ. Microbiol. 1991, 57, 1070–1074. [Google Scholar] [CrossRef]
- Volossiouk, T.; Robb, E.J.; Nazar, R.N. Direct DNA Extraction for PCR-Mediated Assays of Soil Organisms. Appl. Environ. Microbiol. 1995, 61, 3972–3976. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Satyanarayana, T. An Improved Protocol for DNA Extraction from Alkaline Soil and Sediment Samples for Constructing Metagenomic Libraries. Appl. Biochem. Biotechnol. 2011, 165, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Devi, T.; Verma, V.; Rasool, S. Comparative Studies on the Extraction of Metagenomic DNA from Various Soil and Sediment Samples of Jammu and Kashmir Region in Prospect for Novel Biocatalysts. IOSR J. Environ. Sci. Toxicol. Food Technol. 2014, 8, 46–56. [Google Scholar] [CrossRef]
- Martin-Laurent, F.; Philippot, L.; Hallet, S.; Chaussod, R.; Germon, J.C.; Soulas, G.; Catroux, G. DNA Extraction from Soils: Old Bias for New Microbial Diversity Analysis Methods. Appl. Environ. Microbiol. 2001, 67, 4397. [Google Scholar] [CrossRef]
- Petric, I.; Philippot, L.; Abbate, C.; Bispo, A.; Chesnot, T.; Hallin, S.; Laval, K.; Lebeau, T.; Lemanceau, P.; Leyval, C.; et al. Inter-Laboratory Evaluation of the ISO Standard 11063 “Soil Quality—Method to Directly Extract DNA from Soil Samples”. J. Microbiol. Methods 2011, 84, 454–460. [Google Scholar] [CrossRef]
- Técher, D.; Martinez-Chois, C.; D’Innocenzo, M.; Laval-Gilly, P.; Bennasroune, A.; Foucaud, L.; Falla, J. Novel Perspectives to Purify Genomic DNA from High Humic Acid Content and Contaminated Soils. Sep. Purif. Technol. 2010, 75, 81–86. [Google Scholar] [CrossRef]
- Santos, S.S.; Nielsen, T.K.; Hansen, L.H.; Winding, A. Comparison of Three DNA Extraction Methods for Recovery of Soil Protist DNA. J. Microbiol. Methods 2015, 115, 13–19. [Google Scholar] [CrossRef]
- Plassart, P.; Terrat, S.; Thomson, B.; Griffiths, R.; Dequiedt, S.; Lelievre, M.; Regnier, T.; Nowak, V.; Bailey, M.; Lemanceau, P.; et al. Evaluation of the ISO Standard 11063 DNA Extraction Procedure for Assessing Soil Microbial Abundance and Community Structure. PLoS ONE 2012, 7, e44279. [Google Scholar] [CrossRef]
- Basim, Y.; Mohebali, G.; Jorfi, S.; Nabizadeh, R.; Ghadiri, A.; Moghadam, M.A.; Soleymani, F.; Fard, N.J.H. Comparison of Performance and Efficiency of Four Methods to Extract Genomic DNA from Oil Contaminated Soils in Southwestern of Iran. J. Environ. Health Sci. Eng. 2020, 18, 463–468. [Google Scholar] [CrossRef]
- Bollmann-Giolai, A.; Giolai, M.; Heavens, D.; Macaulay, I.; Malone, J.; Clark, M.D. A Low-Cost Pipeline for Soil Microbiome Profiling. Microbiologyopen 2020, 9, e1133. [Google Scholar] [CrossRef]
- Leite, D.C.A.; Balieiro, F.C.; Pires, C.A.; Madari, B.E.; Rosado, A.S.; Coutinho, H.L.C.; Peixoto, R.S. Comparison of DNA Extraction Protocols for Microbial Communities from Soil Treated with Biochar. Braz. J. Microbiol. 2014, 45, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Bürgmann, H.; Pesaro, M.; Widmer, F.; Zeyer, J. A Strategy for Optimizing Quality and Quantity of DNA Extracted from Soil. J. Microbiol. Methods 2001, 45, 7–20. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, K.D.; Yilmaz-Swenson, T.; Elsayed, N.M.; Cruz, D.A.; Rodriguez, R.R.; Kritzer, M.D.; Peterchev, A.V.; Gray, M.; Lewis, S.R.; Roach, J.; et al. Comparative Evaluation of a New Magnetic Bead-Based DNA Extraction Method from Fecal Samples for Downstream next-Generation 16S RRNA Gene Sequencing. PLoS ONE 2018, 13, e0202858. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, M.; Kim, S.W.; Kim, H.S.; Kim, K.Y.; Park, H.B.; Kim, K.J.; Lee, Y.S. Bacterial Community and Diversity from the Watermelon Cultivated Soils through next Generation Sequencing Approach. Plant Pathol. J. 2021, 37, 521–532. [Google Scholar] [CrossRef]
- Wüst, P.K.; Nacke, H.; Kaiser, K.; Marhan, S.; Sikorski, J.; Kandeler, E.; Daniel, R.; Overmann, J. Estimates of Soil Bacterial Ribosome Content and Diversity Are Significantly Affected by the Nucleic Acid Extraction Method Employed. Appl. Environ. Microbiol. 2016, 82, 2595–2607. [Google Scholar] [CrossRef]
- Sagova-Mareckova, M.; Cermak, L.; Novotna, J.; Plhackova, K.; Forstova, J.; Kopecky, J. Innovative Methods for Soil DNA Purification Tested in Soils with Widely Differing Characteristics. Appl. Environ. Microbiol. 2008, 74, 2902–2907. [Google Scholar] [CrossRef]
- Santos, S.S.; Nunes, I.; Nielsen, T.K.; Jacquiod, S.; Hansen, L.H.; Winding, A. Soil DNA Extraction Procedure Influences Protist 18S RRNA Gene Community Profiling Outcome. Protist 2017, 168, 283–293. [Google Scholar] [CrossRef]
- Morita, H.; Akao, S. The Effect of Soil Sample Size, for Practical DNA Extraction, on Soil Microbial Diversity in Different Taxonomic Ranks. PLoS ONE 2021, 16, e0260121. [Google Scholar] [CrossRef]
- Rousk, J.; Brookes, P.C.; Bååth, E. Contrasting Soil PH Effects on Fungal and Bacterial Growth Suggest Functional Redundancy in Carbon Mineralization. Appl. Environ. Microbiol. 2009, 75, 1589–1596. [Google Scholar] [CrossRef]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil Bacterial and Fungal Communities across a PH Gradient in an Arable Soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Guerrieri, A.; Bonin, A.; Münkemüller, T.; Gielly, L.; Thuiller, W.; Francesco Ficetola, G. Effects of Soil Preservation for Biodiversity Monitoring Using Environmental DNA. Mol. Ecol. 2021, 30, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Stach, J.E.M.; Bathe, S.; Clapp, J.P.; Burns, R.G. PCR-SSCP Comparison of 16S RDNA Sequence Diversity in Soil DNA Obtained Using Different Isolation and Purification Methods. FEMS Microbiol. Ecol. 2001, 36, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, L.M.; Sul, W.J.; Blackwood, C.B. Assessment of Bias Associated with Incomplete Extraction of Microbial DNA from Soil. Appl. Environ. Microbiol. 2009, 75, 5428–5433. [Google Scholar] [CrossRef]
- Zielińska, S.; Radkowski, P.; Blendowska, A.; Ludwig-Gałęzowska, A.; Łoś, J.M.; Łoś, M. The Choice of the DNA Extraction Method May Influence the Outcome of the Soil Microbial Community Structure Analysis. MicrobiologyOpen 2017, 6, e00453. [Google Scholar] [CrossRef]
- De Vrieze, J.; Ijaz, U.Z.; Saunders, A.M.; Theuerl, S. Terminal Restriction Fragment Length Polymorphism Is an “Old School” Reliable Technique for Swift Microbial Community Screening in Anaerobic Digestion. Sci. Rep. 2018, 8, 16818. [Google Scholar] [CrossRef] [PubMed]
- Valášková, V.; Baldrian, P. Denaturing Gradient Gel Electrophoresis as a Fingerprinting Method for the Analysis of Soil Microbial Communities. Plant Soil Environ. 2009, 55, 413–423. [Google Scholar] [CrossRef]
- Fajardo, C.; García-Cantalejo, J.; Botías, P.; Costa, G.; Nande, M.; Martin, M. New Insights into the Impact of NZVI on Soil Microbial Biodiversity and Functionality. J. Environ. Sci. Health Part A 2019, 54, 157–167. [Google Scholar] [CrossRef]
- Liu, W.T.; Marsh, T.L.; Cheng, H.; Forney, L.J. Characterization of Microbial Diversity by Determining Terminal Restriction Fragment Length Polymorphisms of Genes Encoding 16S RRNA. Appl. Environ. Microbiol. 1997, 63, 4516–4522. [Google Scholar] [CrossRef]
- Blaud, A.; Diouf, F.; Herrmann, A.M.; Lerch, T.Z. Analysing the Effect of Soil Organic Matter on Bacterial Communities Using T-RFLP Fingerprinting: Different Methods, Different Stories? Biol. Fertil. Soils 2015, 51, 959–971. [Google Scholar] [CrossRef][Green Version]
- van Dorst, J.; Bissett, A.; Palmer, A.S.; Brown, M.; Snape, I.; Stark, J.S.; Raymond, B.; McKinlay, J.; Ji, M.; Winsley, T.; et al. Community Fingerprinting in a Sequencing World. FEMS Microbiol. Ecol. 2014, 89, 316–330. [Google Scholar] [CrossRef]
- Gao, G.; Yin, D.; Chen, S.; Xia, F.; Yang, J.; Li, Q.; Wang, W. Effect of Biocontrol Agent Pseudomonas Fluorescens 2P24 on Soil Fungal Community in Cucumber Rhizosphere Using T-RFLP and DGGE. PLoS ONE 2012, 7, e31806. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, T.; Zhu, L.; Wang, X.; Wang, J.; Wang, J.; Du, Z.; Li, B. Effects of Successive Metalaxyl Application on Soil Microorganisms and the Residue Dynamics. Ecol. Indic. 2019, 103, 194–201. [Google Scholar] [CrossRef]
- Gryta, A.; Frąc, M. Methodological Aspects of Multiplex Terminal Restriction Fragment Length Polymorphism-Technique to Describe the Genetic Diversity of Soil Bacteria, Archaea and Fungi. Sensors 2020, 20, 3292. [Google Scholar] [CrossRef] [PubMed]
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities along an Altitudinal Gradient in Alpine Forest Soils: What Are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef]
- Smith, C.J.; Osborn, A.M. Advantages and Limitations of Quantitative PCR (Q-PCR)-Based Approaches in Microbial Ecology: Application of Q-PCR in Microbial Ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Comparison of Droplet Digital PCR and Quantitative Real-Time PCR for Examining Population Dynamics of Bacteria in Soil. Appl. Microbiol. Biotechnol. 2014, 98, 6105–6113. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hid-dessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Muhae-Ud-Din, G.; Liu, T.; Chen, W.; Zhang, J.; Gao, L. Development of the Droplet Digital PCR to Detect the Teliospores of Tilletia Controversa Kühn in the Soil with Greatly Enhanced Sensitivity. Front. Microbiol. 2020, 11, 4. [Google Scholar] [CrossRef]
- Voegel, T.M.; Larrabee, M.M.; Nelson, L.M. Development of Droplet Digital PCR Assays to Quantify Genes Involved in Nitrification and Denitrification, Comparison with Quantitative Real-Time PCR and Validation of Assays in Vineyard Soil. Can. J. Microbiol. 2021, 67, 174–187. [Google Scholar] [CrossRef]
- Prosser, J.I. Dispersing Misconceptions and Identifying Opportunities for the Use of “omics” in Soil Microbial Ecology. Nat. Rev. Microbiol. 2015, 13, 439–446. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Sáenz, J.S.; Roldan, F.; Junca, H.; Arbeli, Z. Effect of the Extraction and Purification of Soil DNA and Pooling of PCR Amplification Products on the Description of Bacterial and Archaeal Communities. J. Appl. Microbiol. 2019, 126, 1454–1467. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yu, Y.; Li, H.; He, J.; Lee, S.H.; Sun, K. Phylogenetic Diversity of Planktonic Bacteria in the Chukchi Borderland Region in Summer. Hai Yang Xue Bao 2013, 32, 66–74. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The Bioinformatics Tools for the Genome Assembly and Analysis Based on Third-Generation Sequencing. Brief. Funct. Genom. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Patrick, K. 454 Life Sciences: Illuminating the Future of Genome Sequencing and Personalized Medicine. Yale J. Biol. Med. 2007, 80, 191–194. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Illumina Sequencing Technology Highest Data Accuracy, Simple Workflow, and a Broad Range of Applications. Available online: https://www.illumina.com/documents/products/techspotlights/techspotlight_sequencing.pdf (accessed on 23 October 2022).
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S RRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The Impact of Next-Generation Sequencing on Genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Can, T. Introduction to Bioinformatics. In miRNomics: MicroRNA Biology and Computational Analysis; Humana Press: Totowa, NJ, USA, 2014; pp. 51–71. [Google Scholar]
- Lesk, A. Introduction to Bioinformatics, 5th ed.; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Ejigu, G.F.; Jung, J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology 2020, 9, 295. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Hupfauf, S.; Etemadi, M.; Fernández-Delgado Juárez, M.; Gómez-Brandón, M.; Insam, H.; Podmirseg, S.M. CoMA—An Intuitive and User-Friendly Pipeline for Amplicon-Sequencing Data Analysis. PLoS ONE 2020, 15, e0243241. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing Bioinformatic Pipelines for Microbial 16S RRNA Amplicon Sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef]
- Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining Operational Taxonomic Units Using DNA Barcode Data. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1935–1943. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-Throughput Amplicon Sequencing of the Full-Length 16S RRNA Gene with Single-Nucleotide Resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [PubMed]
- Semenov, M.V.; Krasnov, G.S.; Semenov, V.M.; van Bruggen, A.H.C. Long-Term Fertilization Rather than Plant Species Shapes Rhizosphere and Bulk Soil Prokaryotic Communities in Agroecosystems. Appl. Soil Ecol. 2020, 154, 103641. [Google Scholar] [CrossRef]
- de la Fuente, G.; Belanche, A.; Girwood, S.E.; Pinloche, E.; Wilkinson, T.; Newbold, C.J. Pros and Cons of Ion-Torrent next Generation Sequencing versus Terminal Restriction Fragment Length Polymorphism T-RFLP for Studying the Rumen Bacterial Community. PLoS ONE 2014, 9, e101435. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M data management and analysis system v.6.0: New tools and advanced capabilities. Nucleic Acids Res. 2020, 49, D751–D763. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Beisel, J.-N.; Usseglio-Polatera, P.; Bachmann, V.; Moreteau, J.-C. A Comparative Analysis of Evenness Index Sensitivity. Int. Rev. Hydrobiol. 2003, 88, 3–15. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-Parametric Multivariate Analyses of Changes in Community Structure. Austral. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- ANOSIM: Analysis of Similarities. Available online: https://rdrr.io/rforge/vegan/man/anosim.html (accessed on 19 October 2022).
- Principal Coordinate Analysis and Non-Metric Multidimensional Scaling. In Statistics for Biology and Health; Springer: New York, NY, USA, 2007; pp. 259–264.
- Anderson, M.J.; Willis, T.J. Canonical Analysis of Principal Coordinates: A Useful Method of Constrained Ordination for Ecology. Ecology 2003, 84, 511–525. [Google Scholar] [CrossRef]
- Gryta, A.; Frąc, M.; Oszust, K. Community Shift in Structure and Functions across Soil Profile in Response to Organic Waste and Mineral Fertilization Strategies. Appl. Soil Ecol. 2019, 143, 55–60. [Google Scholar] [CrossRef]
- Ondreičková, K.; Gubišová, M.; Piliarová, M.; Horník, M.; Matušinský, P.; Gubiš, J.; Klčová, L.; Hudcovicová, M.; Kraic, J. Responses of Rhizosphere Fungal Communities to the Sewage Sludge Application into the Soil. Microorganisms 2019, 7, 505. [Google Scholar] [CrossRef] [PubMed]
- Čuhel, J.; Malý, S.; Královec, J. Shifts and recovery of soil microbial communities in a 40-year field trial under mineral fertilization. Pedobiologia 2019, 77, 150575. [Google Scholar] [CrossRef]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel Production Related to Soil Microbial Diversity and Evenness. Microbiol. Spectr. 2021, 9, e0022921. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Liu, R.; Li, L.; Wang, L.; Wang, W. Insight into Bacterial Community Diversity and Monthly Fluctuations of Medicago Sativa Rhizosphere Soil in Response to Hydrogen Gas Using Illumina High-Throughput Sequencing. Curr. Microbiol. 2018, 75, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, F.; Hu, C.; Liu, B. Metagenomics Reveals Taxon-Specific Responses of the Nitrogen-Cycling Microbial Community to Long-Term Nitrogen Fertilization. Soil Biol. Biochem. 2021, 156, 108214. [Google Scholar] [CrossRef]
- Akinola, S.A.; Ayangbenro, A.S.; Babalola, O.O. The Diverse Functional Genes of Maize Rhizosphere Microbiota Assessed Using Shotgun Metagenomics. J. Sci. Food Agric. 2021, 101, 3193–3201. [Google Scholar] [CrossRef]
- Sadet-Bourgeteau, S.; Houot, S.; Karimi, B.; Mathieu, O.; Mercier, V.; Montenach, D.; Morvan, T.; Sappin-Didier, V.; Watteau, F.; Nowak, V.; et al. Microbial Communities from Different Soil Types Respond Differently to Organic Waste Input. Appl. Soil Ecol. 2019, 143, 70–79. [Google Scholar] [CrossRef]
- Wydro, U.; Jabłońska-Trypuć, A.; Hawrylik, E.; Butarewicz, A.; Rodziewicz, J.; Janczukowicz, W.; Wołejko, E. Heavy Metals Behavior in Soil/Plant System after Sewage Sludge Application. Energies 2021, 14, 1584. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Z.; Hu, S.; Ruan, Z.; Jiang, J.; Chen, C.; Shen, Z. Response of Soil Bacterial Communities to Lead and Zinc Pollution Revealed by Illumina MiSeq Sequencing Investigation. Environ. Sci. Pollut. Res. Int. 2017, 24, 666–675. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, J.; Lu, J.; Sun, Y. Study on the Influence of Soil Microbial Community on the Long-Term Heavy Metal Pollution of Different Land Use Types and Depth Layers in Mine. Ecotoxicol. Environ. Saf. 2019, 170, 218–226. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, W.; Xu, H.; Cui, X.; Li, J.; Chen, J.; Zheng, B. Characterizations of Heavy Metal Contamination, Microbial Community, and Resistance Genes in a Tailing of the Largest Copper Mine in China. Environ. Pollut. 2021, 280, 116947. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, X.; Liu, J.; Cheng, Y.; Zou, J.; Zhai, F.; Sun, Z.; Han, L. Soil Microbial Community Succession and Interactions during Combined Plant/White-Rot Fungus Remediation of Polycyclic Aromatic Hydrocarbons. Sci. Total Environ. 2021, 752, 142224. [Google Scholar] [CrossRef] [PubMed]
- Dou, R.; Sun, J.; Lu, J.; Deng, F.; Yang, C.; Lu, G.; Dang, Z. Bacterial Communities and Functional Genes Stimulated during Phenanthrene Degradation in Soil by Bio-Microcapsules. Ecotoxicol. Environ. Saf. 2021, 212, 111970. [Google Scholar] [CrossRef] [PubMed]
- Łozowicka, B.; Wołejko, E.; Kaczyński, P.; Konecki, R.; Iwaniuk, P.; Drągowski, W.; Łozowicki, J.; Tujtebajeva, G.; Wydro, U.; Jablońska-Trypuć, A. Effect of Microorganism on Behaviour of Two Commonly Used Herbicides in Wheat/Soil System. Appl. Soil Ecol. 2021, 162, 103879. [Google Scholar] [CrossRef]
- Singh, A.K.; Singla, P. Biodegradation of Diuron by Endophytic Bacillus Licheniformis Strain SDS12 and Its Application in Reducing Diuron Toxicity for Green Algae. Environ. Sci. Pollut. Res. Int. 2019, 26, 26972–26981. [Google Scholar] [CrossRef]
- Serbent, M.P.; Dos Anjos Borges, L.G.; Quadros, A.; Marconatto, L.; Tavares, L.B.B.; Giongo, A. Prokaryotic and Microeukaryotic Communities in an Experimental Rice Plantation under Long-Term Use of Pesticides. Environ. Sci. Pollut. Res. Int. 2021, 28, 2328–2341. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| References | Compared Procedures | Amount of DNA | Purity of DNA A260/280/A260/230 | Comments |
|---|---|---|---|---|
| [42] | manual lysozyme/phenol–chloroform method | 430.85 ng/µL | 1.665/1.24 | Incubation with lysozyme at 60 °C for 1 h, addition of RNase at 37 °C for 30 min and SDS |
| Qiagen DNeasy Blood & Tissue Kit | 561.3 ng/µL | 1.915/1.55 | sample was incubated at 75 °C for 10 min, silica-column based adsorption of DNA | |
| QIAamp DNA Stool mini kit | 76.35 ng/µL | 1.66/0.97 | sample was incubated at 75 °C for 10 min, silica-column based adsorption of DNA | |
| [36] | Polyethylene Glycol (PEG)/NaCl Method | 0.73 µg/mL | 1.26/1.12 | Lysis with liquid nitrogen; not suitable for PCR amplification for fungal study |
| Soil Master DNA extraction kit; | 0.79 µg/ mL | 1.32/1.21 | Lysis with liquid nitrogen; not suitable for PCR amplification for fungal study | |
| mannitol-PBS-PEG/NaCl | 2.20 µg/ mL | 1.81 /1.84 | recovery of high molecular weight soil DNA | |
| Mannitol-PBS-PCI | 2.36 µg/ mL | 1.84/1.93 | recovery of high molecular weight soil DNA | |
| Mannitol-PBS-CTAB | 2.67 µg/ mL | 1.85/2.07 | recovery of high molecular weight soil DNA | |
| [8] | Tsai and Olson method [43] | 7.55 μg/g of soil | 1.18/0.82 | physical cell lysis by repeated freeze–thaw cycles |
| Volossiouk et al. method (1995) [44] | 9.36 μg/g of soil | 1.11/0.85 | physical cell lysis by crushing soil in liquid nitrogen with freeze–thaw process; extraction cells with SDS-phenol and collected by ethanol precipitation | |
| Extraction with CTAB buffer [32] | 19.1 μg/g of soil | 1.25/0.94 | CTAB extraction buffer method; successful extraction of high-molecular weight metagenomic DNA | |
| Verma and Satyanarayana method [45] | 11.23 μg/g of soil | 1.48/1.32 | focused on cell lysis and DNA purification steps | |
| Singh et al. method [46] | 1.33 μg/g of soil | 1.02/1.00 | Extraction with Chloroform: Isoamyl alcohol (24:1); DNA precipitation with 50% PEG and 0.1 volume of 1M NaCl at −20 °C for 1h; removal of humic using CTAB extraction buffer, DNA precipitation by PEG/NaCl followed by DNA purification using 2% (w/v) CaCl2 solution; extraction of high-molecular weight, | |
| improved method | 15.55 μg/g of soil | 1.74/1.70 | using of enzymatic (lysozyme and proteinase K) and chemical (CTAB and CaCl2) strategies for cell lysis; precipitation with PEG (polyethylene glycol) and isopropanol | |
| [11] | Martin-Laurent et al.method [47] with modification by Petric et al. method [48] | 1.51–3.42 μg/g dry soil | 1.48–1.79/0.21–0.36 | Using glass beads of different sizes (300 mg of 106-μm-diameter glass beads and 2 of 2 mm diameter); 10 min incubation with potassium acetate (protein removing) |
| Técher et al. method [49] | 2.23–10.70 μg/g dry soil | 0.86–4.72/0.50–0.68 | Sand of different sizes (500 mg of fine sand and 200 mg of quartz sand); vitamin mix was used to humic acids precipitation | |
| [37] | NucleoSpin® soil kit | 0.73–2.54 μg/g of soil | 0.43–0.83/1.30–1.59 | Contain two different lysis buffers to optimize the extraction efficiency according to the soil properties, bead-beating(ceramic beads) |
| innuSPEED soil DNA kit | n.d | n.d | Lysis: Lysis tube B (2 mL) heat treatment (95–98 °C, 20 min), mechanical lysis via glass beads, 600 μL buffer; failed to extract DNA | |
| FastDNA® SPIN kit for soil | 0.36–1.34 μg/g of soil | 0.03–0.24/1.12–1.50 | Lysis tube E (2 mL), mechanical lysis via matrix E (1·4 mm ceramic spheres, 0·1 mm silica spheres and one 4 mm glass bead) | |
| [50] | Power Lyzer™ PowerSoil® DNA Isolation Kit | 8.7–47.5 μg/g of soil | 1.8–1.9/1.5–2.1 | - |
| the ISO standardmethod modified by Plassart et al. [51] | 21.5–43.4 μg/g of soil | 1.5/1.6–1.8 | Include mechanical lysis step; | |
| GnS-GII protocol | 8.2–49.7 μg/g of soil | 1.6–1.7/1.5–1.6 | Lysis with glass beads in different sizes and with freeze–thaw; precipitation in icecold isopropanol, humic removing by two different minicolumns | |
| Protocol A | 10 µg/mL | 1.9/2.4 | homogenization buffer consist of 100 mm Tris HCL, 100 mm EDTA, 1.5 M NaCl and 30 mg lysozyme | |
| [52] | Protocol B | 14 µg/mL | 1.6/0.65 | three-step soil washing |
| Soil DNA extraction kit, (MACHEREY-NAGEL) | 14 µg/mL | 2.2/0.86 | - | |
| Protocol D | 135 µg/mL | 2/2.2 | utilizing of physical (silica beads, freeze and melt) and chemical lysis simultaneously on both sediments (from primary stage and after homogenization buffer) | |
| [53] | PowerSoil® DNA Isolation Kit | 0–1203 ng | 2.02–2.12/0.82–1.77 | silica column-based DNA extraction with extensive washing |
| FastDNA™ SPIN Kit for Soil | 1914–20,333 ng | 1.26–1.87/0.06–0.35 | uses a large amount of binding material | |
| SDE method | 468–2913 | 1.29–1.31/0.60–0.87 | Lysis with 1 mm diameter garnet particles and three metal 4 mm bearings, removal humic acids with aluminum sulfate; gDNA purification with magnetic beads |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wydro, U. Soil Microbiome Study Based on DNA Extraction: A Review. Water 2022, 14, 3999. https://doi.org/10.3390/w14243999
Wydro U. Soil Microbiome Study Based on DNA Extraction: A Review. Water. 2022; 14(24):3999. https://doi.org/10.3390/w14243999
Chicago/Turabian StyleWydro, Urszula. 2022. "Soil Microbiome Study Based on DNA Extraction: A Review" Water 14, no. 24: 3999. https://doi.org/10.3390/w14243999
APA StyleWydro, U. (2022). Soil Microbiome Study Based on DNA Extraction: A Review. Water, 14(24), 3999. https://doi.org/10.3390/w14243999