
Passive Sampling with Active Carbon Fibres in the Determination of Organic Pollutants in Groundwater

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Preparation
2.2.1. ACF Purification and Transport to Sampling Site
2.2.2. ACF Deployment
2.2.3. ACF Collection and Elution of Compounds
2.2.4. Elution of Compounds from ACFs
2.3. Analytical Method–Chromatographic Analysis
2.4. Quality Control
2.4.1. Sampling QC Procedure
2.4.2. Analytical QC Procedure
2.5. Validation of Method
2.6. Data Sets of Presented Examples
3. Results
3.1. Design and Use of ACF Passive Samplers in Groundwater
3.2. Optimisation of Purification, Elution, and Desorption Procedure from Active Carbon Fibres
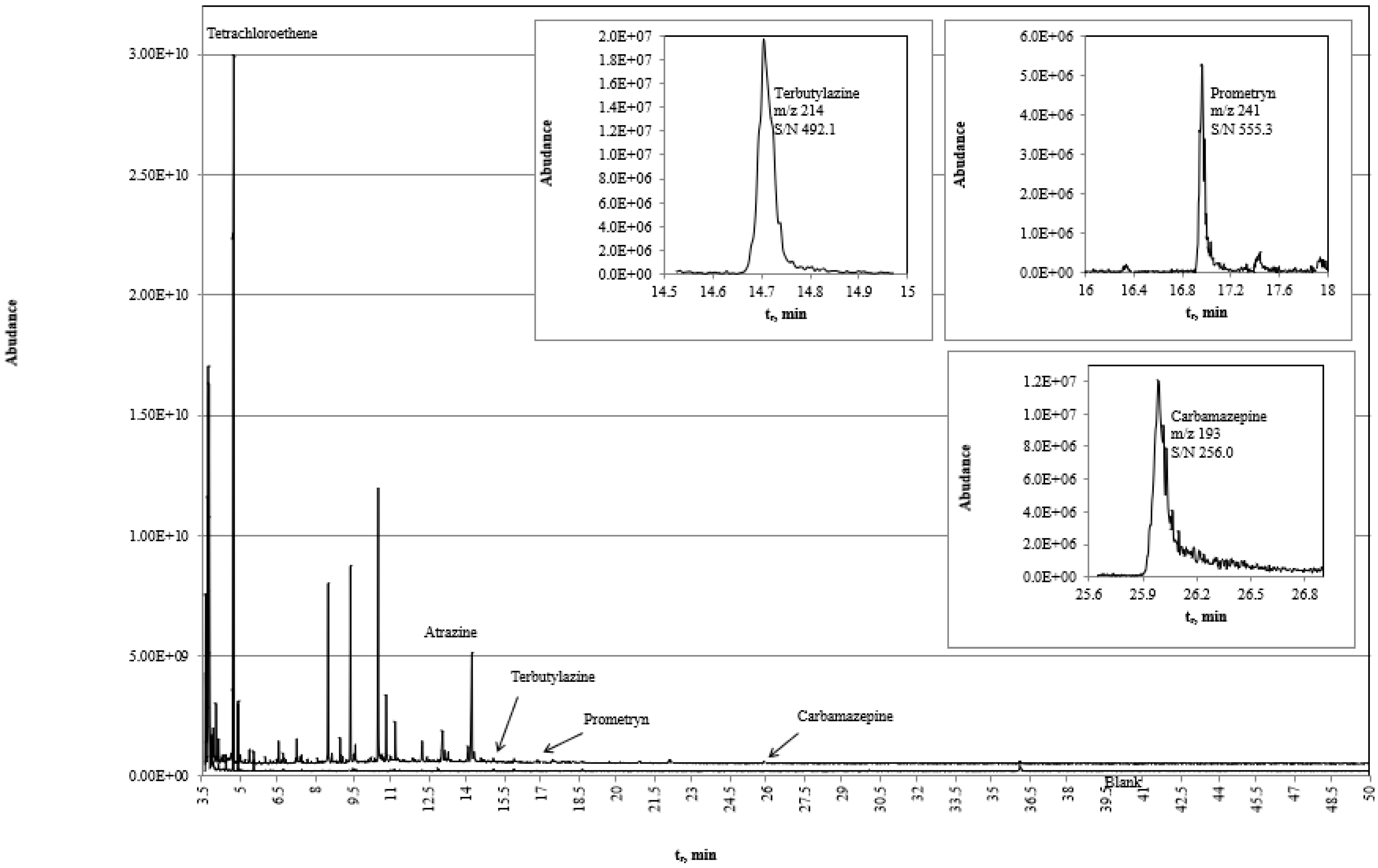
3.3. Confirmation of Non-Targeted Screening
3.4. Influence of Exposure Time in Highly Polluted Groundwater
3.5. Validation on Synthetic Samples
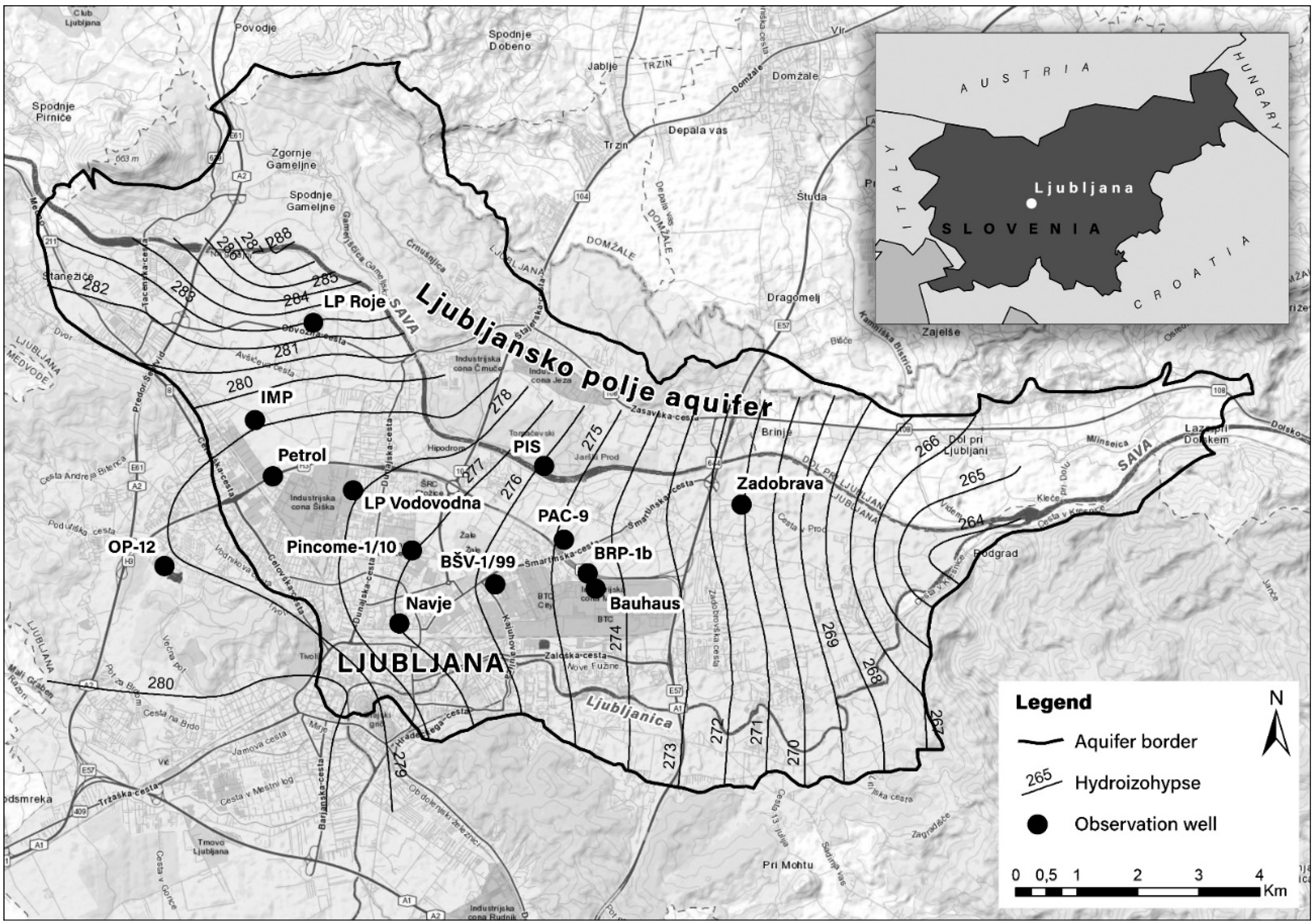
3.6. Applicability of the AFC Method in Groundwater Quality Studies
3.6.1. Example 1—Non-Targeted Qualitative Screening
3.6.2. Example 2—Identifying the Presence and Source of Organic Compounds in Groundwater
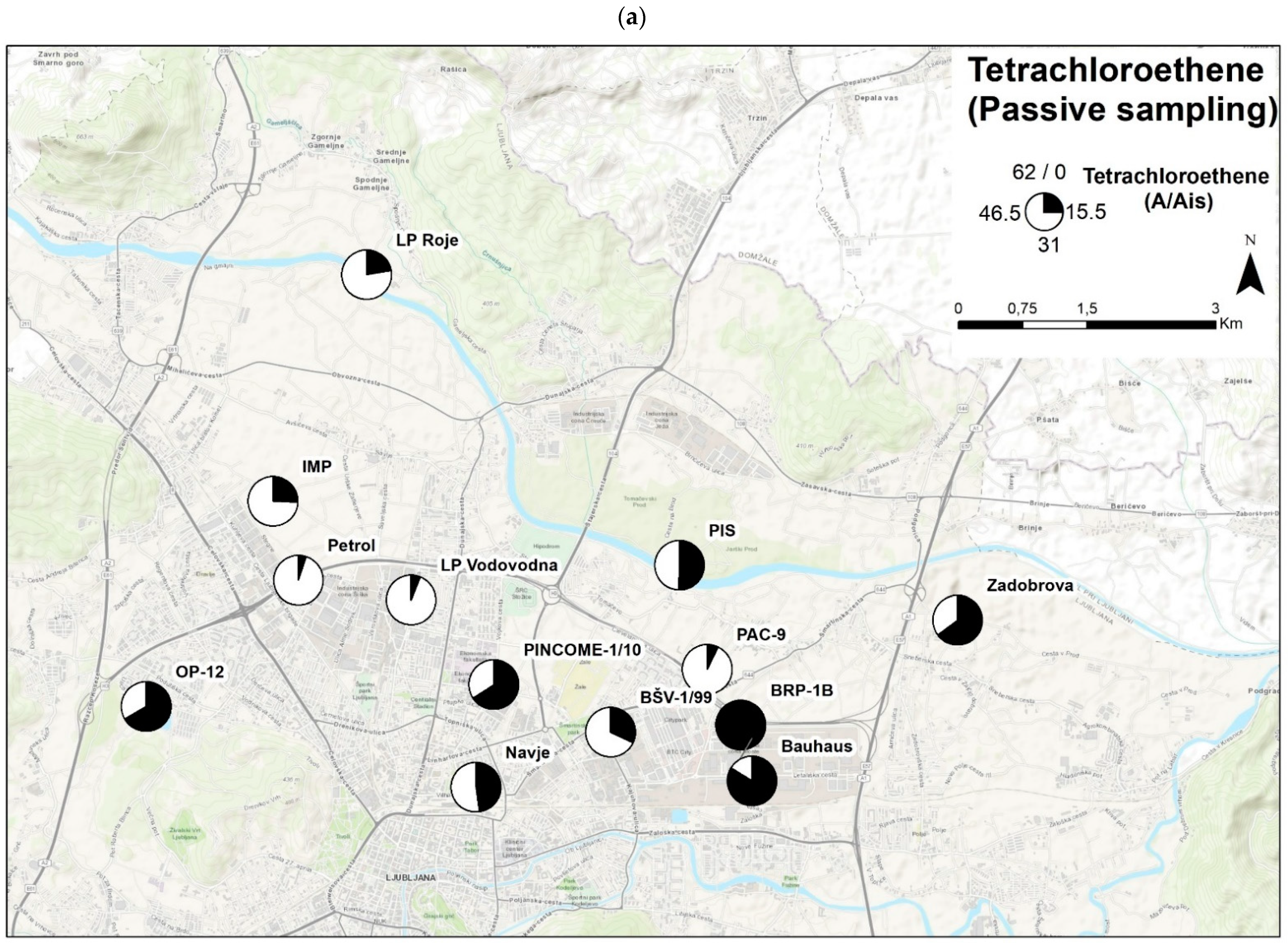
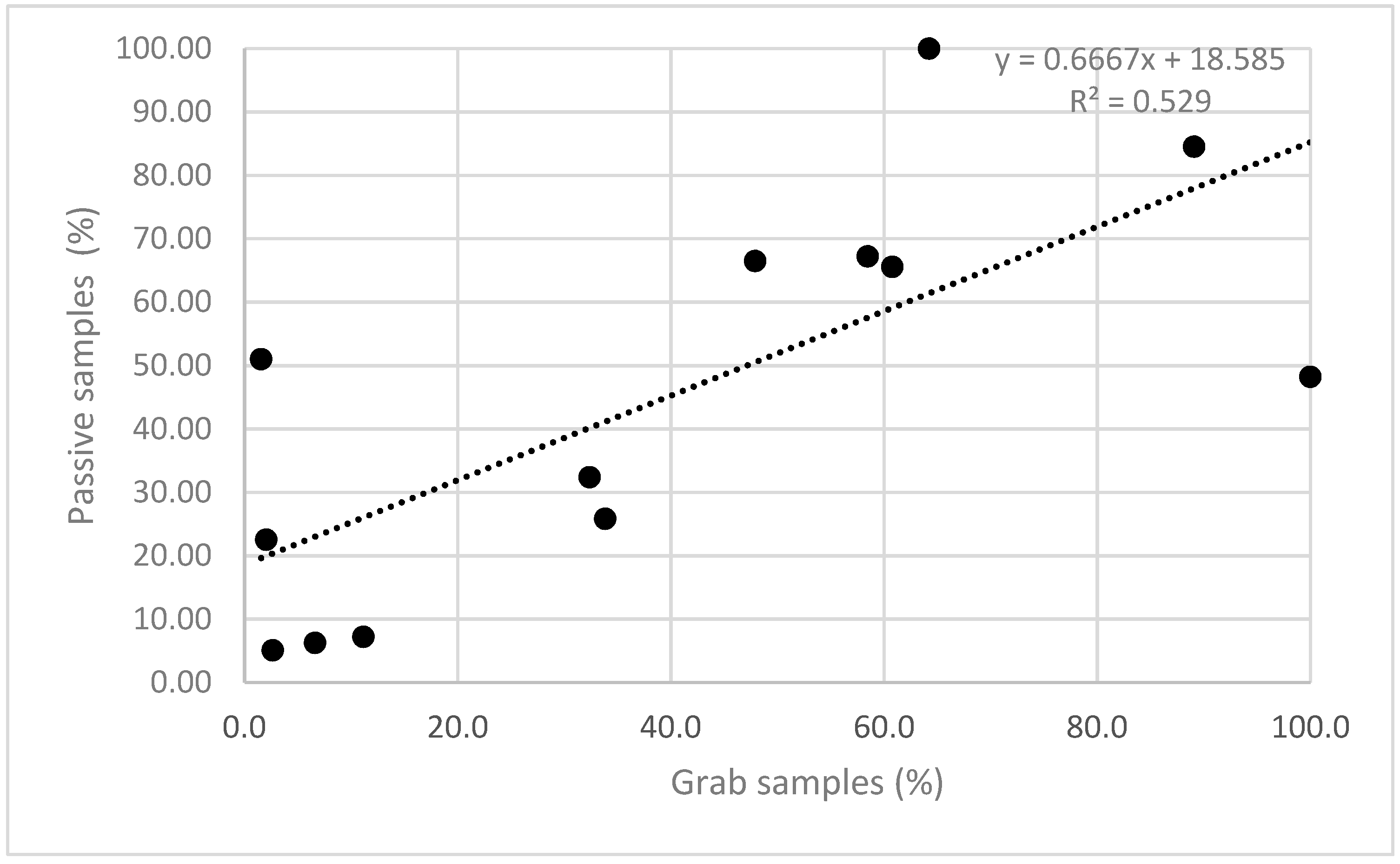
3.6.3. Example 3—Comparison of the Passive Sampling Method with the Grab Samples
4. Discussion
- (1)
- This paper also demonstrates the advantage and applicability of using ACF passive samplers for organic compound screening in groundwater. More organic compounds were detected and identified with ACF passive samplers than through the analysis of grab samples. The sampling method is capable of detecting a wide range of organic compounds unselectively in a single shot.
- (2)
- In the case of groundwater samples from all over Slovenia, 892 organic compounds were detected with ACF passive samplers. It has been proven that this sampling is capable of detecting a wide range of compounds unselectively. In this process, the presence of various organic contaminants in groundwater was identified. Through the use of the AFC sampling technique, we were able to detect a wide range of previously unknown and unspecified compounds in groundwater. This methodology also revealed the presence of transformation products, one of which had not been previously identified at the sampling sites. The results (an extensive list of compounds) are important for the design of different schemes used for monitoring groundwater quality and have the ability to also prioritise the less well-known compounds in groundwater.
- (3)
- In the case of the Ljubljansko polje aquifer, it has been shown that the identification of organic compounds in groundwater may serve to help assess the risk of potential anthropogenic contamination. This method allows us to detect and evaluate the presence of pollutants and identify their anthropogenic source. From the results of passive sampling, we were able to identify the main compounds and determine their typical use and origin. Depending on the type of compound, we can determine whether the potential contamination of groundwater is agricultural, urban, or industrial in origin.
- (4)
- Parallels between the results of the analysis with passive samplers and the quantitative chemical analysis at the Ljubljansko polje aquifer indicate that the passive sampler method detects the presence of a compound in groundwater already at the level of pg/L. This means that with the passive sampler method, we are able to detect the presence of very low concentrations of certain compounds in the groundwater that cannot yet be routinely detected using quantitative chemical analyses.
- (5)
- ACF passive samplers are useful for long-term deployments (3 months or more) and for temporal as well as spatial assessments of groundwater concentrations. The effect of the displacement of particular compounds due to more strongly adsorbed compounds was not considered relevant under the circumstances.
- (6)
- A field validation was conducted at sampling sites, and a comparison of samples between passive samplers and samples from quantitative chemical analysis was performed based on normalised values. Results show a good overlapping of results for PCE (R2 = 0.53). Spatial comparison of PCE distribution throughout the Ljubljansko polje aquifer also showed minor deviations, which might have occurred due to the difference in the time frame of grab and passive sampling.
- (7)
- The method for groundwater monitoring with passive sampling introduced and optimized herein can be used in a wide range of research projects and monitoring campaigns and is comparable to other passive sampling techniques.
- (8)
- Further investigation of the ACF passive samplers’ performance on validation techniques and the evaluation of uncertainty would be appropriate. Moreover, to test for a broader applicability of ACF passive samplers, investigations into their longer deployments under different physicochemical conditions and in additional waters, such as surface and wastewater, would be beneficial.
- (9)
- The method could be very efficient for screening difficult-to-reach areas by interested parties due to the simple field equipment and also undisturbed long-distance transport of samples to the laboratory enabled by the stability of ACFs and the efficient prevention and complete control of possible contamination of samples during their installation and transport. In this case, consideration should be given to the possibility of a trained sampling instructor accessing the sampling site remotely during each sampling.
- (10)
- Last but not least, data collected from groundwater samples obtained using the passive sampling technique could be used for various types of multivariate statistical modelling to identify sources of pollution in the environment. Some studies on the use of passive samplers have already been published in the literature [17,75].
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Locatelli, M.; Sciasci, F.; Cifelli, R.; Malatesta, L.; Bruni, P.; Croce, F. Analytical methods for the endocrine disruptor compounds determination in environmental water samples. J. Chromatogr. A 2016, 1434, 1–18. [Google Scholar] [CrossRef]
- Mirasole, C.; Di Carro, M.; Tanwar, S.; Magi, E. Liquid chromatography–tandem mass spectrometry and passive sampling: Powerful tools for the determination of emerging pollutants in water for human consumption. J. Mass Spectrom. 2016, 51, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Vrana, B.; Allan, I.J.; Greenwood, R.; Mills, G.A.; Dominiak, E.; Svensson, K.; Knutsson, J.; Morrison, G. Passive sampling techniques for monitoring pollutants in water. TrAC Trends Anal. Chem. 2005, 24, 845–868. [Google Scholar] [CrossRef]
- Allan, I.J.; Harman, C.; Ranneklev, S.B.; Thomas, K.V.; Grung, M. Passive sampling for target and nontarget analyses of moderately polar and nonpolar substances in water. Environ. Toxicol. Chem. 2013, 32, 1718–1726. [Google Scholar] [CrossRef]
- Lindholm, P.C.; Knuutinen, J.S.; Ahkola, H.S.J.; Herve, S.H. Analysis of trace pharmaceuticals and related compounds in municipal wastewaters by preconcentration, chromatography, derivatisation, and separation methods. BioResources 2014, 9, 3688–3732. [Google Scholar] [CrossRef]
- Allinson, G.; Allinson, M.; Kadokami, K. Combining passive sampling with a GC-MS-database screening tool to assess trace organic contamination of rivers: A pilot study in Melbourne, Australia. Water Air Soil Pollut. 2015, 226, 230. [Google Scholar] [CrossRef]
- Koroša, A.; Auersperger, P.; Mali, N. Determination of micro-organic contaminants in groundwater (Maribor, Slovenia). Sci. Total Environ. 2016, 571, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Soulier, C.; Coureau, C.; Togola, A. Environmental forensics in groundwater coupling passive sampling and high resolution mass spectrometry for screening. Sci. Total Environ. 2016, 563–564, 845–854. [Google Scholar] [CrossRef]
- Auersperger, P.; Lah, K.; Vrbec, A.; Zidar, V.K.; Koroša, A.; Mali, N. Tracing pollutants in groundwater by passive sampling and gas chromatography mass spectrometry. In Proceedings of the 31st International Symposium on Chromatography, ISC-16, Cork, Ireland, 28 August–1 September 2016. [Google Scholar]
- Li, Z.; Sobek, A.; Radke, M. Fate of pharmaceuticals and their transformation products in four small european rivers receiving treated wastewater. Environ. Sci. Technol. 2016, 50, 5614–5621. [Google Scholar] [CrossRef] [PubMed]
- Booij, K.; Robinson, C.D.; Burgess, R.M.; Mayer, P.; Roberts, C.A.; Ahrens, L.; Allan, I.J.; Brant, J.; Jones, L.; Kraus, U.R.; et al. Passive sampling in regulatory chemical monitoring of nonpolar organic compounds in the aquatic environment. Environ. Sci. Technol. 2016, 50, 3–17. [Google Scholar] [CrossRef]
- Terzopoulou, E.; Voutsa, D. Active and passive sampling for the assessment of hydrophilic organic contaminants in a river basin-ecotoxicological risk assessment. Environ. Sci. Pollut. Res. Int. 2016, 23, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Křesinová, Z.; Petrů, K.; Lhotský, O.; Rodsand, T.; Cajthaml, T. Passive sampling of pharmaceuticals and personal care products in aquatic environments. Eur. J. Environ. Sci. 2016, 6, 43–56. [Google Scholar] [CrossRef][Green Version]
- Corada-Fernández, C.; Candela, L.; Torres-Fuentes, N.; Pintado-Herrera, M.G.; Paniw, M.; González-Mazo, E. Effects of extreme rainfall events on the distribution of selected emerging contaminants in surface and groundwater: The Guadalete River basin (SW, Spain). Sci. Total Environ. 2017, 605–606, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Mali, N.; Cerar, S.; Koroša, A.; Auersperger, P. Passive sampling as a tool for identifying micro-organic compounds in groundwater. Sci. Total Environ. 2017, 593–594, 722–734. [Google Scholar] [CrossRef]
- Lapworth, D.J.; Lopez, B.; Laabs, V.; Kozel, R.; Wolter, R.; Ward, R.; Amelin, E.V.; Besien, T.; Claessens, J.; Delloye, F.; et al. Developing a groundwater watch list for substances of emerging concern: A European perspective. Environ. Res. Lett. 2018, 14, 035004. [Google Scholar] [CrossRef]
- Pinasseau, L.; Wiest, L.; Fildier, A.; Volatier, L.; Fones, G.R.; Mills, G.A.; Mermillod-Blondin, F.; Vulliet, E. Use of passive sampling and high resolution mass spectrometry using a suspect screening approach to characterise emerging pollutants in contaminated groundwater and runoff. Sci. Total Environ. 2019, 672, 253–263. [Google Scholar] [CrossRef]
- Kiefer, K.; Müller, A.; Singer, H.; Hollender, J. New relevant pesticide transformation products in groundwater detected using target and suspect screening for agricultural and urban micropollutants with LC-HRMS. Water Res. 2019, 165, 114972. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S.; et al. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef] [PubMed]
- Trullols, E.; Ruisánchez, I.; Rius, F.X. Validation of qualitative analytical methods. TrAC Trends Anal. Chem. 2004, 23, 137–145. [Google Scholar] [CrossRef]
- Velcárcel, M.; Cárdenas, S.; Gallego, M. Qualitative analysis revisited. Crit. Rev. Anal. Chem. 2000, 30, 345–361. [Google Scholar] [CrossRef]
- De Brabander, H.F.; Batjoens, P.; De Wasch, K.; Courtheyn, D.; Pottie, G.; Smets, F. Qualitative or quantitative methods for residue analysis? TrAC Trends Anal. Chem. 1997, 16, 485–489. [Google Scholar] [CrossRef]
- Muñoz-Olivas, R. Screening analysis: An overview of methods applied to environmental, clinical and food analyses. TrAC Trends Anal. Chem. 2004, 23, 203–216. [Google Scholar] [CrossRef]
- Simonet, B.M.; Ríos, A.; Valcárcel, M. Unreliability of screening methods. Anal. Chimica Acta 2004, 516, 67–74. [Google Scholar] [CrossRef]
- Ellison, S.L.R.; Gregory, S. PerspectiveQuantifying uncertainty in qualitative analysis. Analyst 1998, 123, 1155–1161. [Google Scholar] [CrossRef]
- Mil’man, B.L.; Konopel’ko, L.A. Uncertainty of qualitative chemical analysis: General methodology and binary test methods. J. Anal. Chem. 2004, 59, 1128–1141. [Google Scholar] [CrossRef]
- Pulido, A.; Ruisánchez, I.; Boqué, R.; Rius, F.X. Estimating the uncertainty of binary test results to assess their compliance with regulatory limits. Anal. Chimica Acta 2002, 455, 267–275. [Google Scholar] [CrossRef]
- Pulido, A.; Ruisánchez, I.; Boqué, R.; Rius, F.X. Uncertainty of results in routine qualitative analysis. TrAC Trends Anal. Chem. 2003, 22, 647–654. [Google Scholar] [CrossRef]
- Valcárcel, M.; Cárdenas, S. Current and future screening systems. Anal. Bioanal. Chem. 2004, 381, 81–83. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed. Eurachem. 2014. Available online: https://www.eurachem.org.xn--ivg (accessed on 19 December 2021).
- Alvarez, D.A.; Petty, J.D.; Huckins, J.N.; Jones-Lepp, T.L.; Getting, D.T.; Goddard, J.P.; Manahan, S.E. Development of a passive, in situ, integrative sampler for hydrophilic organic contaminants in aquatic environments. Environ. Toxicol. Chem. 2004, 23, 1640–1648. [Google Scholar] [CrossRef]
- Van Metre, P.C.; Alvarez, D.; Mahler, B.J.; Nowell, L.; Sandstrom, M.; Moran, P. Complex mixtures of pesticides in Midwest, U.S. streams indicated by POCIS time-integrating samplers. Environ. Pollut. 2017, 220, 431–440. [Google Scholar] [CrossRef]
- Charriau, A.; Lissalde, S.; Poulier, G.; Mazzella, N.; Buzier, R.; Guibaud, G. Overview of the Chemcatcher® for the passive sampling of various pollutants in aquatic environments Part A: Principles, calibration, preparation and analysis of the sampler. Talanta 2016, 148, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Lissalde, S.; Charriau, A.; Poulier, G.; Mazzella, N.; Buzier, R.; Guibaud, G. Overview of the Chemcatcher® for the passive sampling of various pollutants in aquatic environments Part B: Field handling and environmental applications for the monitoring of pollutants and their biological effects. Talanta 2016, 148, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Petrie, B.; Gravell, A.; Mills, G.A.; Youdan, J.; Barden, R.; Kasprzyk-Hordern, B. In situ calibration of a new chemcatcher configuration for the determination of polar organic micropollutants in wastewater effluent. Environ. Sci. Technol. 2016, 50, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Vermeirssen, E.L.M.; Bramaz, N.; Hollender, J.; Singer, H.; Escher, B.I. Passive sampling combined with ecotoxicological and chemical analysis of pharmaceuticals and biocides—Evaluation of three Chemcatcher™ configurations. Water Res. 2009, 43, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, E.F.; Menezes, H.C.; Cardeal, Z.L. New passive sampling device for effective monitoring of pesticides in water. Anal. Chim. Acta 2019, 1054, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, E.F.; Menezes, H.C.; Cardeal, Z.L. Passive and grab sampling methods to assess pesticide residues in water. A review. Environ. Chem. Lett. 2020, 18, 1019–1048. [Google Scholar] [CrossRef]
- Taylor, A.C.; Fones, G.R.; Mills, G.A. Trends in the use of passive sampling for monitoring polar pesticides in water. Trends Environ. Anal. Chem. 2020, 27, e00096. [Google Scholar] [CrossRef]
- Brielmann, H.; Kralik, M.; Humer, F.; Clara, M.; Weiss, S.; Kulscar, S.; Scharf, S.; Voerkelius, S. Identifying anthropogenic nitrogen sources in ground and surface water. In Proceedings of the International Symposium on Isotope Hydrology: Revisiting Foundations and Exploring Frontiers, Vienna, Austria, 11–15 May 2015. [Google Scholar]
- Ahrens, L.; Daneshvar, A.; Lau, A.E.; Kreuger, J. Characterization of five passive sampling devices for monitoring of pesticides in water. J. Chromatogr. A 2015, 1405, 1–11. [Google Scholar] [CrossRef]
- Verreydt, G.; Bronders, J.; Keer, I.V. Lab and field screening of 5 selected passive samplers for the measurement of VOC fluxes in groundwater. J. Agric. Sci. Appl. 2014, 3, 30–38. [Google Scholar] [CrossRef]
- Togola, A.; Berho, C.; Bruchet, A.; Robert, S. POCIS for pesticide monitoring in groundwaters: From “low flow” lab calibration to in situ monitoring. In Proceedings of the SETAC Europe 25th Annual Meeting, Barcelona, Spain, 3–7 May 2015. [Google Scholar]
- McHugh, T.E.; Kulkarni, P.R.; Newell, C.J.; Britt, S.L. Methods for Minimization and Management of Variability in Long Term Groundwater Monitoring Results; Technical Report, No. ER-201209; U.S. Department of Defense: Washington, DC, USA, 2015; p. 50.
- Roll, I.B.; Halden, R.U. Critical review of factors governing data quality of integrative samplers employed in environmental water monitoring. Water Res. 2016, 94, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sophie, L.-F.; Brieudes, V.; Lalere, B.; Candido, P.; Couturier, G.; Budzinski, H.; Lavison-Bompard, G. For more reliable measurements of pharmaceuticals in the environment: Overall measurement uncertainty estimation, QA/QC implementation and metrological considerations. A case study on the Seine River. TrAC Trends Anal. Chem. 2016, 77, 76–86. [Google Scholar]
- Criquet, J.; Dumoulin, D.; Howsam, M.; Mondamert, L.; Goossens, J.-F.; Prygiel, J.; Billon, G. Comparison of POCIS passive samplers vs. composite water sampling: A case study. Sci. Total Environ. 2017, 609, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Kaserzon, S.L.; Vijayasarathy, S.; Bräunig, J.; Mueller, L.; Hawker, D.W.; Thomas, K.V.; Mueller, J. Calibration and validation of a novel passive sampling device for the time integrative monitoring of per- and polyfluoroalkyl substances (PFASs) and precursors in contaminated groundwater. J. Hazard. Mater. 2018, 366, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Pinasseau, L.; Wiest, L.; Volatier, L.; Fones, G.R.; Mills, G.A.; Mermillod-Blondin, F.; Vulliet, E. Calibration and field application of an innovative passive sampler for monitoring groundwater quality. Talanta 2019, 208, 120307. [Google Scholar] [CrossRef] [PubMed]
- Musa Abubakar, T.; Amimul, A.; Abubakar, S.; Moetaz, E.; Arunkumar, T.; Bipin, J.; Razzaque, M.; Norsyahariati, N. A review on activated carbon: Process, application and prospects. J. Adv. Civ. Eng. Pract. Res. 2016, 2, 7–13. [Google Scholar]
- Boopathy, R.; Sekar, K.; Asit, M.; Ganesan, S. Adsorption of ammonium ion by coconut shell-activated carbon from aqueous solution: Kinetic, isotherm, and thermodynamic studies. Environ. Sci. Pollut. Res. 2012, 20, 533–542. [Google Scholar] [CrossRef]
- Rivera, J.; Ventura, F.; Caixach, J.; De Torres, M.; Figueras, A.; Guardiola, J. GC/MS, HPLC and FAB mass spectrometric analysis of organic micropollutants in Barcelona’s water supply. Int. J. Environ. Anal. Chem. 1987, 29, 15–35. [Google Scholar] [CrossRef]
- Kadokami, K.; Koga, M.; Otsuki, A. Gas chromatography-mass spectrometric determination of traces of hydrophilic and volatile organic compounds in water after preconcentration with activated carbon. Anal. Sci. 1990, 6, 843–849. [Google Scholar] [CrossRef]
- Seethapathy, S.; Górecki, T.; Li, X. Passive sampling in environmental analysis. J. Chromatogr. A 2008, 1184, 234–253. [Google Scholar] [CrossRef]
- Hale, E.S.; Tomaszewski, E.J.; Luthy, G.R.; Werner, D. Sorption of dichlorodiphenyltrichloroethane (DDT) and its metabolites by activated carbon in clean water and sediment slurries. Water Res. 2009, 43, 4336–4346. [Google Scholar] [CrossRef]
- Nyoni, H.; Chimuka, L.; Vrana, B.; Cukrowska, E. Membrane assisted passive sampler for triazine compounds in water bodies—Characterization of environmental conditions and field performance. Anal. Chim. Acta 2011, 694, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.D.; Beddows, P.A.; Bouchot, G.G.; Metcalfe, T.L.; Li, H.; Van Lavieren, H. Contaminants in the coastal karst aquifer system along the Caribbean coast of the Yucatan Peninsula, Mexico. Environ. Pollut. 2011, 159, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Tapie, N.; LeMenacha, K.; Pasquaudb, S.; Elieb, P.; Deviera, M.H.; Budzinski, H. PBDE and PCB contamination of eels from the Gironde estuary: From glass eels to silver eels. Chemosphere 2011, 83, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Berho, C.; Togola, A.; Coureau, C.; Ghestem, J.P.; Amalric, L. Applicability of polar organic compound integrative samplers for monitoring pesticides in groundwater. Environ. Sci. Pollut. Res. 2013, 20, 5220–5228. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.; Togola, A.; Gonzalez, C. In-situ calibration of POCIS for the sampling of polar pesticides and metabolites in surface water. Talanta 2013, 116, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Vrana, B.; Klucarova, V.; Benicka, E.; Abou-Mrad, N.; Amdany, R.; Horakova, S.; Draxler, A.; Humer, F.; Gans, O. Passive sampling: An effective method for monitoring seasonal and spatial variability of dissolved hydrophobic organic contaminants and metals in the Danube River. Environ. Pollut. 2014, 184, 101–112. [Google Scholar] [CrossRef]
- Silvani, L.; Riccardi, C.; Eek, E.; Papini, M.P.; Morin, N.A.; Cornelissen, G.; Oen, A.M.; Hale, S.E. Monitoring alkylphenols in water using the polar organic chemical integrative sampler (POCIS): Determining sampling rates via the extraction of PES membranes and Oasis beads. Chemosphere 2017, 184, 1362–1371. [Google Scholar] [CrossRef]
- Morin, N.A.O.; Mazzella, N.; Arp, H.P.H.; Randon, J.; Camilleri, J.; Wiest, L.; Coquery, M.; Miège, C. Kinetic accumulation processes and models for 43 micropollutants in “pharmaceutical” POCIS. Sci. Total Environ. 2018, 615, 197–207. [Google Scholar] [CrossRef]
- Stuart, M.; Lapworth, D.; Crane, E.; Hart, A. Review of risk from potential emerging contaminants in UK groundwater. Sci. Total Environ. 2012, 416, 1–21. [Google Scholar] [CrossRef]
- Manamsa, K.; Crane, E.; Stuart, M.; Talbot, J.; Lapworth, D.; Hart, A. A national-scale assessment of micro-organic contaminants in groundwater of England and Wales. Sci. Total Environ. 2016, 568, 712–726. [Google Scholar] [CrossRef]
- Shareef, A.; Parnis, C.J.; Angove, M.J.; Wells, J.D.; Johnson, B.B. Suitability of N,O-bis(trimethylsilyl)trifluoroacetamide and N-(tert-butyldimethylsilyl)-N methyltrifluoroacetamide as derivatisation reagents for the determination of the estrogens estrone and 17α-ethinylestradiol by gas chromatography–mass spectrometry. J. Chromatogr. A 2004, 10, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Auersperger, P.; Lah, K.; Kus, J.; Marsel, J. High precision procedure for determination of selected herbicides and their degradation products in drinking water by solid-phase extraction and gas chromatography–mass spectrometry. J. Chromatogr. A 2005, 1088, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Žlebnik, L. Pleistocene deposits of the Kranj, Sora, and Ljubljana fields. Geologija 1971, 14, 5–51. (In Slovenian) [Google Scholar]
- Drobne, F.; Mencej, Z.; Brilly, M. Preveritve in Dopolnitve Strokovnih Osnov za Določitev Varstvenih Pasov Sedanjih in Perspektivnih Vodnih Virov za Območje Mesta Ljubljane in Okolice; Delovno Poročilo Inštituta za Geologijo, Geotehniko in Geofiziko: Ljubljana, Slovenia, 1997. [Google Scholar]
- Urbanc, J.; Jamnik, B. Porazdelitev in izvor nitratov v podzemni vodi ljubljanskega polja. Geologija 2007, 50, 467–475. [Google Scholar] [CrossRef]
- Jamnik, B.; Urbanc, J. Origin and guality of groundwater from Ljubljansko polje. RMZ Mater. Geoenviron. 2000, 47, 167–178. [Google Scholar]
- Postigo, C.; Martinez, D.E.; Grondona, S.; Miglioranza, K.S.B. Groundwater pollution: Sources, mechanisms, and prevention A2. In Encyclopedia of the Anthropocene; Dellasala, D.A., Goldstein, M.I., Eds.; Elsevier: Oxford, UK, 2018; pp. 87–96. [Google Scholar]
- European Commission (EC). Commission decision of 10 March 2004 concerning the non-inclusion of atrazine in Annex I to Council Directive 91/414/EEC and the withdrawal of authorisations for plant protection products containing this active substance 2004/247/EC. Off. J. Eur. Union. 2004, 248, 50–55. [Google Scholar]
- Jamnik, B.; Žitnik, M. Letno Poročilo o Skladnosti Pitne Vode na Oskrbovalnem Območju v Upravljanju Javnega Podjetja Vodovod-Kanalizacija d.o.o. v Letu 2017; Javno Podjetje Vodovod-Kanalizacija d.o.o.: Ljubljana, Slovenia, 2018. [Google Scholar]
- Trček, B.; Žigon, D.; Kramarič Zidar, V.; Auersperger, P. The fate of benzotriazole pollutants in an urban oxic intergranular aquifer. Water Res. 2018, 131, 264–273. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CAS NO | tr, min | m/z | Intensity Estimate | A (avg) | RDS (%) | % in AKPV/A(max.)blank |
|---|---|---|---|---|---|---|---|
| (QVN/QLN) | (1–5) | (for QVN m/z) | |||||
| Benzene | 71-43-2 | 2.5 | 78/51 | 3 | 25,220,508 | 34 | 5.9 |
| 1,2,4-trichlorobenzene | 120-82-1 | 8.3 | 180/145,109 | 5 | 18,051,177 | 34 | 0.022 |
| Chlorotoluron | 15545-48-9 (28479-22-3) | 8.8 | 167/132,104 | 2 | 1,152,915 | 51 | 0.11 |
| (as 3-chloro-4-methylphenylisocyanate) | |||||||
| 1-methyl-1H-benzotriazole | 13351-73-0 | 10.3 | 133/10,590 | 5 | 29,498,504 | 47 | 0.072 |
| Atrazine | 1912-24-9 | 14 | 200/215,173 | 3 | 8,552,177 | 54 | 0.015 |
| Caffeine | 58-08-2 | 16 | 194/10,967 | 3 | 5,557,887 | 54 | 0.22 |
| Propyphenazone | 479-92-5 | 17.5 | 215/230 | 3 | 20,360,628 | 68 | 0.078 |
| Carbamazepine | 298-46-4 | 25.9 | 193/236,165 | 3 | 196,786 | 73 | N.D.* |
| Estrone | 53-16-7 | 32.5 | 270/185,146 | 2 | 435,79 | 85 | N.D.* |
| Compound | CAS NO | tr, min | m/z | Intensity Estimate |
|---|---|---|---|---|
| (QVN/QLN) | (1–5) | |||
| 4-t-octylfenol-TBDMS | 140-66-9 | 16.1 | 249/32,073 | 3 |
| 4-nonylphenol-TBDMS | 104-40-5 | 21.6 | 277/334,165 | 2 |
| diclofenac-TBDMS | 15307-86-5 | 30.2 | 352/21,475 | 2 |
| 17beta-estradiol-TBDMS* | 50-28-2 | 38.8 | 329/386,163 | 2 |
| 17alfa-etinylestradiol-TBDMS* | 57-63-6 | 39.8 | 353/410,327 | 2 |
| Parameter/Date | 28 February 2017 | 21 March 2017 | 6 April 2017 | Unit |
|---|---|---|---|---|
| Temperature (on field) | 11.4 | 10.8 | 10.7 | °C |
| Electroconductivity (20 °C) | 658 | 630 | 676 | µS/cm |
| Dissolved oxygen | - | 0.12 | 0.12 | mg/L |
| Total organic carbon | 9.67 | 10.27 | 9.76 | mg/L |
| Ammonium | 7.9 | 7.4 | 7.9 | mg/L |
| Nitrate | <0.2 | - | <0.2 | mg/L |
| Chloride | 28.9 | - | 28.6 | mg/L |
| Hydrogencarbonate | 431 | - | 434 | mg/L |
| Iron, dissolved | - | 3.03 | 2.91 | mg/L |
| Terbutryn | 2.25 | 2.73 | - | µg/L |
| Propyphenazone | 0.072 | 0.087 | - | µg/L |
| Prometryn | 0.11 | <LOD = 0.0020 | - | µg/L |
| Carbamazepine | 0.36 | 0.35 | - | µg/L |
| CAS NO | tr (min) | Substance | Source | Group | Use | Nr. of Detection | % of Detection |
|---|---|---|---|---|---|---|---|
| 127-18-4 | 4.5 | Tetrachloroetene (c.i.) | Dry cleaning, solvent, degreasing in the metal industry | Halogenated solvents | I | 43 | 91.5 |
| 79-01-6 | 3.8 | Trichloroethylene (t.i.) | Dry cleaning, solvent, degreasing in the metal industry | Halogenated solvents | I | 42 | 89.4 |
| 1912-24-9 | 14.2 | Atrazine (c.i.) | Herbicide | Pesticide | A | 40 | 85.1 |
| 6190-65-4 | 13 | Desethylatrazine (c.i.) | Atrazine degradation product | Pesticide | A | 31 | 66.0 |
| 122-34-9 | 14.1 | Simazine (t.i.) | Herbicide | Pesticide | A | 26 | 55.3 |
| - | 9.4 | 2,4-dimethyl-2H-benzotriazole (t.i.) | Degradation of corrosion inhibitors e.g. 4-methyl-1H-benzotriazole | Other Industrial | U | 24 | 51.1 |
| 30125-63-4 | 13.2 | Desethylterbuthylazine (c.i.) | Terbuthylazine degradation product | Pesticide | A | 24 | 51.1 |
| 16584-00-2 | 8.5 | 2-methyl-2H-benzotriazole (c.i.) | Degradation of corrosion inhibitors e.g. 1H-benzotriazole | Other Industrial | U | 22 | 46.8 |
| 427-77-0 | 20.8 | Gibberellin A9 (t.i.) | Natural fungicide | Pesticide | A | 18 | 38.3 |
| 139-40-2 | 14.3 | Propazine (c.i.) | Herbicide | Pesticide | A | 18 | 38.3 |
| 71-55-6 | 3.6 | 1,1,1-Trichloroethane (t.i.) | Solvent | Halogenated solvents | I | 17 | 36.2 |
| 99982-48-6 | 24.7 | Metabolite of Nifedipine (t.i.) | From drug nifedipine | Domestic and personal | U | 15 | 31.9 |
| 5915-41-3 | 14.6 | Terbuthylazine (c.i.) | Herbicide | Pesticide | A | 15 | 31.9 |
| 13674-84-5 | 15 | Tri-(2-chloroisopropyl) phosphate (t.i.) | Flame retardant | Plasticisers and aditives | I | 15 | 31.9 |
| 7287-19-6 | 16.9 | Prometryn (c.i.) | Herbicide | Pesticide | A | 13 | 27.7 |
| 78-40-0 | 7.6 | Triethyl phosphate (t.i.) | Plasticizers | Plasticisers and aditives | I | 13 | 27.7 |
| 314-40-9 | 17.5 | Bromacil (t.i.) | Herbicide | Pesticide | A | 11 | 23.4 |
| 10233-13-3 | 12.5 | Isopropyl laurate (t.i.) | Natural compound, cosmetics | Domestic and personal | U | 11 | 23.4 |
| 298-46-4 | 26 | Carbamazepine (c.i.) | Drug | Domestic and personal | U | 11 | 23.4 |
| - | 5.4 | Unknown compound m/z 31,61 (t.i.) | - | - | U | 11 | 23.4 |
| 112-49-2 | 8.8 | Triethylene Glycol Dimethyl Ether (t.i.) | Solvent | Non-halogenated solvents | I | 10 | 21.3 |
| - | 10.8 | 1,4-dimethyl-1H-benzotriazole (t.i.) | In connection with 2,4-dimethyl-2H-benzotriazole | Other Industrial | U | 9 | 19.1 |
| 20189-42-8 | 8.8 | 3-Ethyl-4-methyl-1H-pyrrole-2,5-dione (t.i.) | Natural compound, green tea, pyrolysis of natural materials, waste water | Domestic and personal | U | 9 | 19.1 |
| 29878-31-7 | 11.7 | 4-methyl-1H-benzotriazole (t.i.) | Corrosion inhibitor, tolytriazole | Domestic and personal | U | 9 | 19.1 |
| 5176-82-9 | 8.6 | 1,3-dimethyl-2,4,5-imidazolidinetrione (t.i.) | Metabolite of caffeine | Domestic and personal | U | 9 | 19.1 |
| 293-30-1 | 8.6 | 1,3,5,7-Tetroxocane (t.i.) | Formaldehide derivative | Domestic and personal | U | 8 | 17.0 |
| 108-38-3 | 5.3 | m- + p-xylene (c.i.) | Automotive, solvent | Non-halogenated solvents | I | 8 | 17.0 |
| 95-47-6 | 5.4 | o-xylene (c.i.) | Automotive, solvent | Non-halogenated solvents | I | 8 | 17.0 |
| Type and Group | Source | Use | n | Share (%) |
|---|---|---|---|---|
| Halogenated solvents | Industrial compounds | I | 156 | 32 |
| Non-halogenated solvents | ||||
| Plasticisers and aditives | ||||
| Domestic and personal | Urban compounds | U | 138 | 28 |
| Other Industrial | ||||
| Pesticides | Agricultural compounds | A | 196 | 40 |
| Sampling Point | Grab Samples (µg/L) | Passive Samples (A/Ais) |
|---|---|---|
| Tetrachloroethene | Tetrachloroethene | |
| LP Roje | <LOD | 13.83 |
| LP Vodovodna | 0.07 | 3.81 |
| PINCOME-1/10 | 0.52 | 40.84 |
| Bauhaus | 0.97 | 51.94 |
| BRP-1B | 0.70 | 61.44 |
| BŠV-1/99 | 0.35 | 19.89 |
| IMP | 0.37 | 15.85 |
| Navje | 1.10 | 29.62 |
| OP-12 | 0.63 | 41.30 |
| PAC-9 | 0.12 | 4.40 |
| Petrol | <LOD = 0.06 | 3.09 |
| PIS | <LOD = 0.06 | 31.30 |
| Zadobrova | 0.66 | 40.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auersperger, P.; Koroša, A.; Mali, N.; Jamnik, B. Passive Sampling with Active Carbon Fibres in the Determination of Organic Pollutants in Groundwater. Water 2022, 14, 585. https://doi.org/10.3390/w14040585
Auersperger P, Koroša A, Mali N, Jamnik B. Passive Sampling with Active Carbon Fibres in the Determination of Organic Pollutants in Groundwater. Water. 2022; 14(4):585. https://doi.org/10.3390/w14040585
Chicago/Turabian StyleAuersperger, Primož, Anja Koroša, Nina Mali, and Brigita Jamnik. 2022. "Passive Sampling with Active Carbon Fibres in the Determination of Organic Pollutants in Groundwater" Water 14, no. 4: 585. https://doi.org/10.3390/w14040585
APA StyleAuersperger, P., Koroša, A., Mali, N., & Jamnik, B. (2022). Passive Sampling with Active Carbon Fibres in the Determination of Organic Pollutants in Groundwater. Water, 14(4), 585. https://doi.org/10.3390/w14040585