
Compositional Changes and Co-Occurrence Patterns of Planktonic Bacteria and Microeukaryotes in a Subtropical Estuarine Ecosystem, the Pearl River Delta


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Sampling
2.3. Chemical Analyses
2.4. Molecular Methods
2.5. Bioinformatics
3. Results
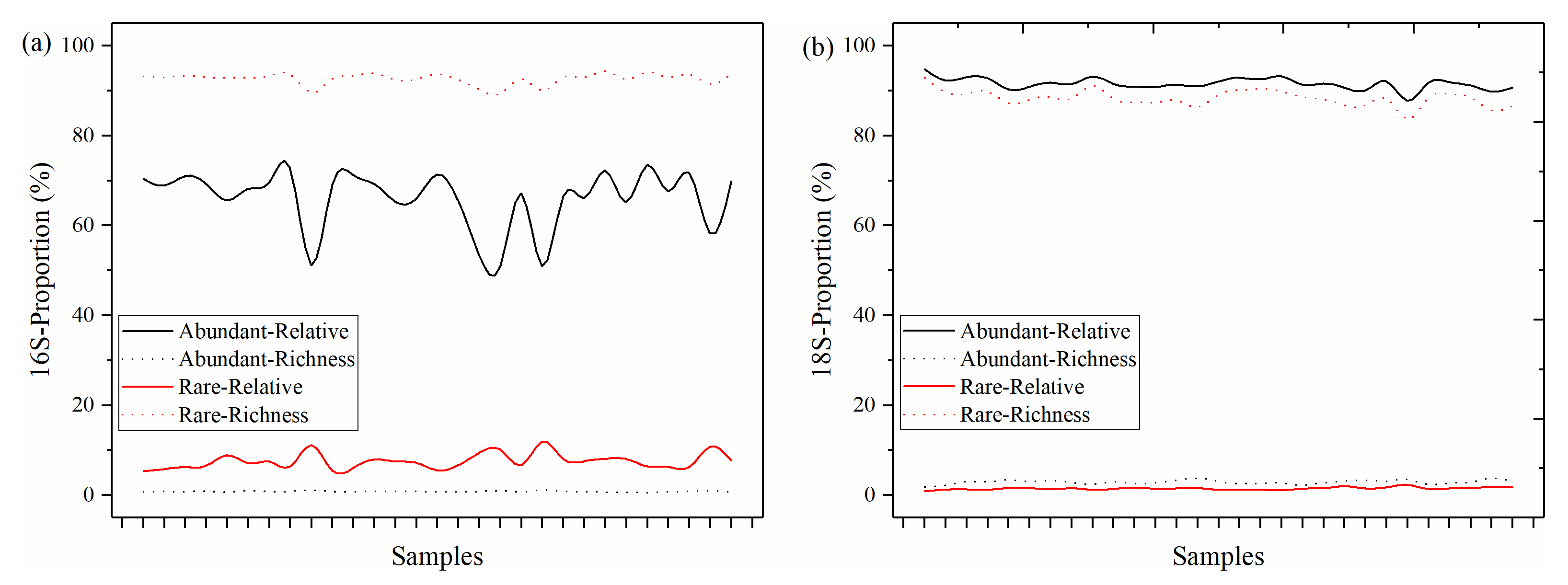
3.1. The Distributions of Abundant and Rare Taxa of Bacteria and Microeukaryotes
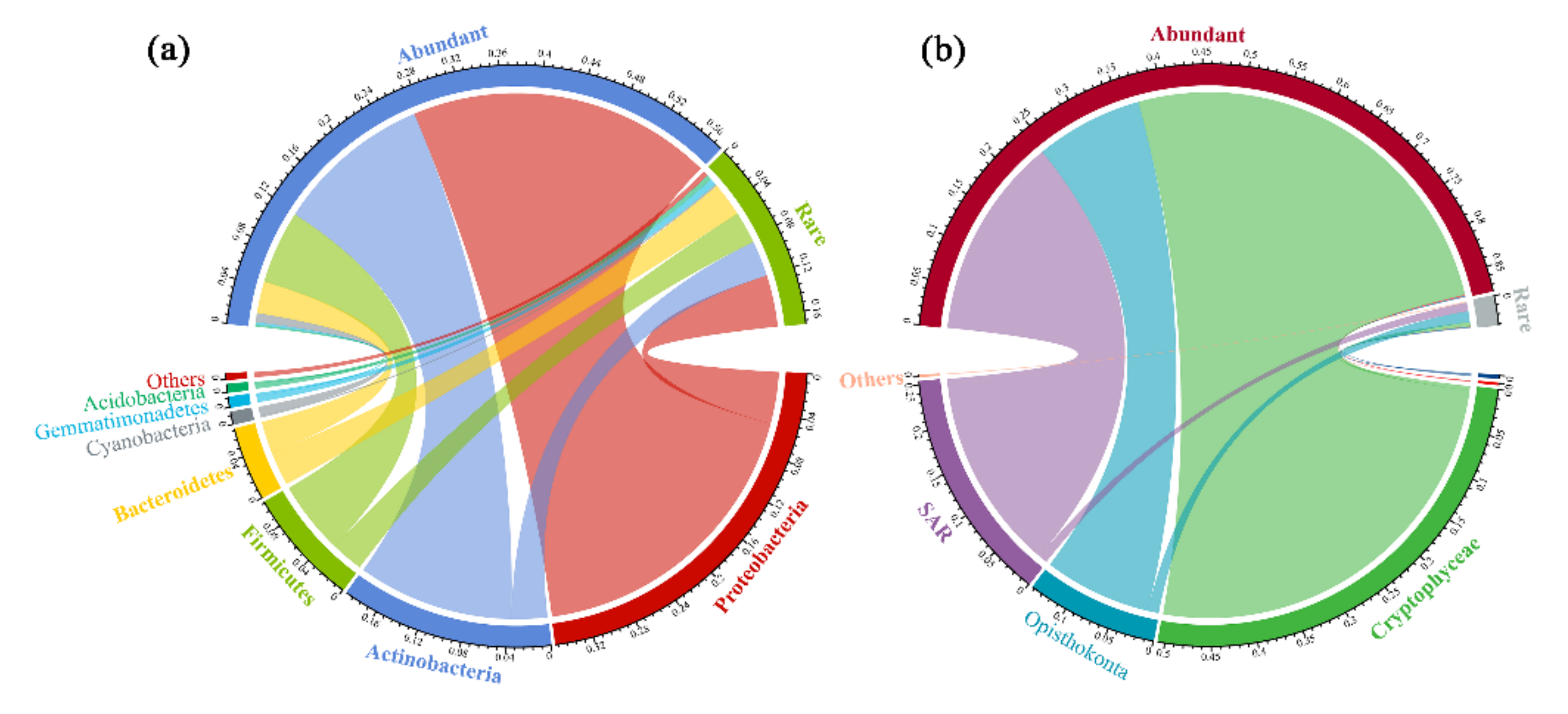
3.2. Taxonomic Compositions of the Abundant and Rare Subcommunities
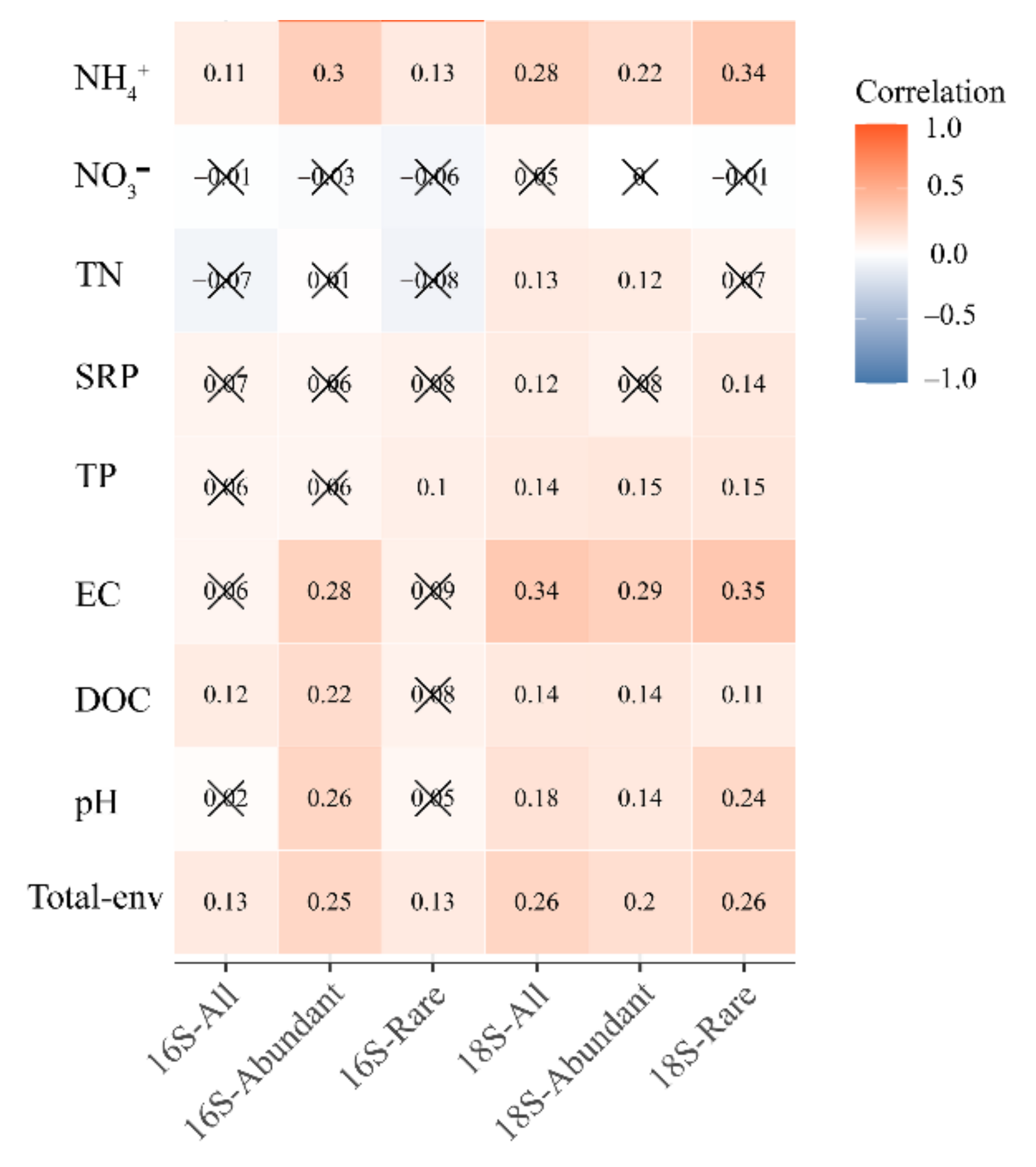
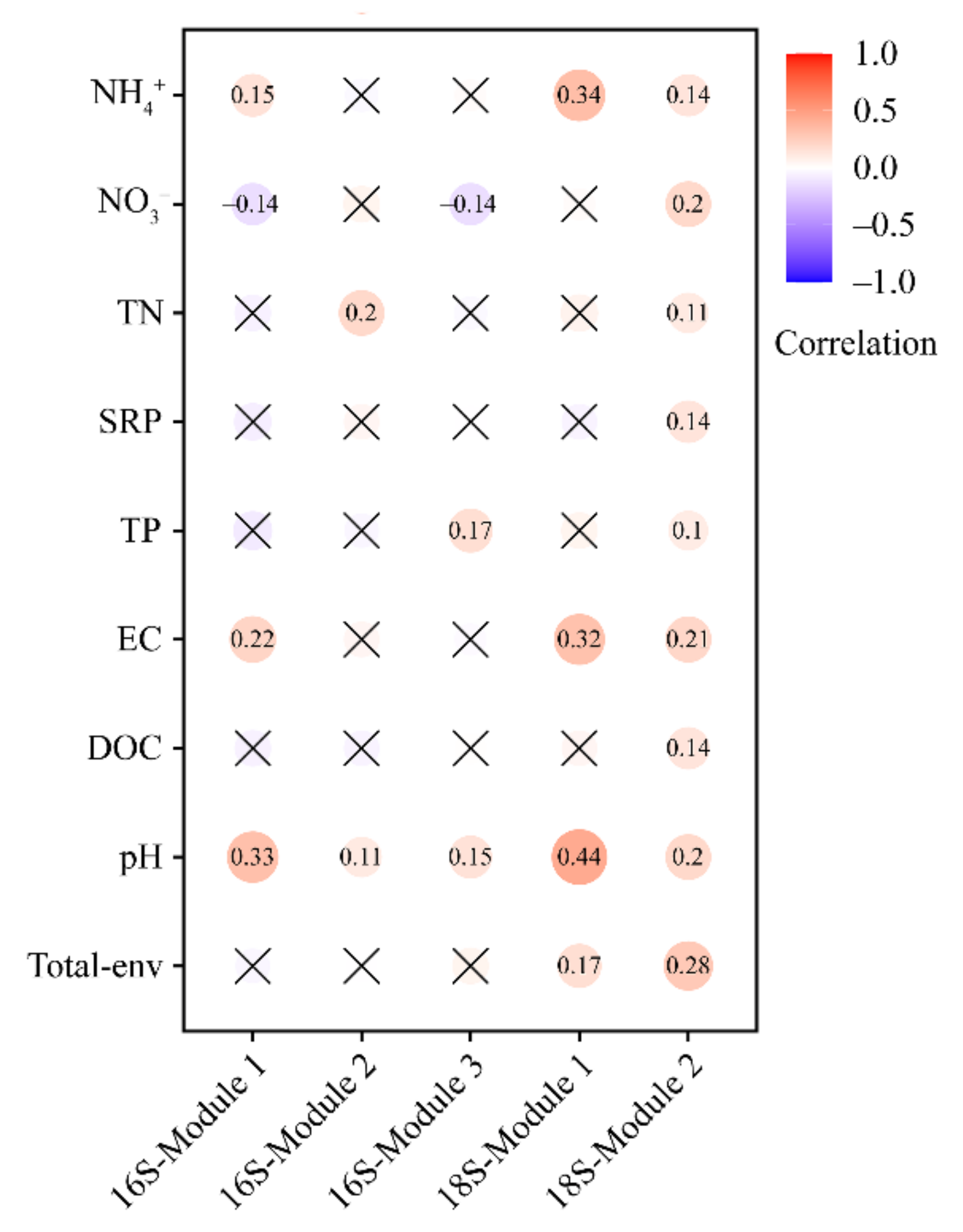
3.3. Environmental Variables Related to the Bacterial and Microeukaryotic Communities and Subcommunities
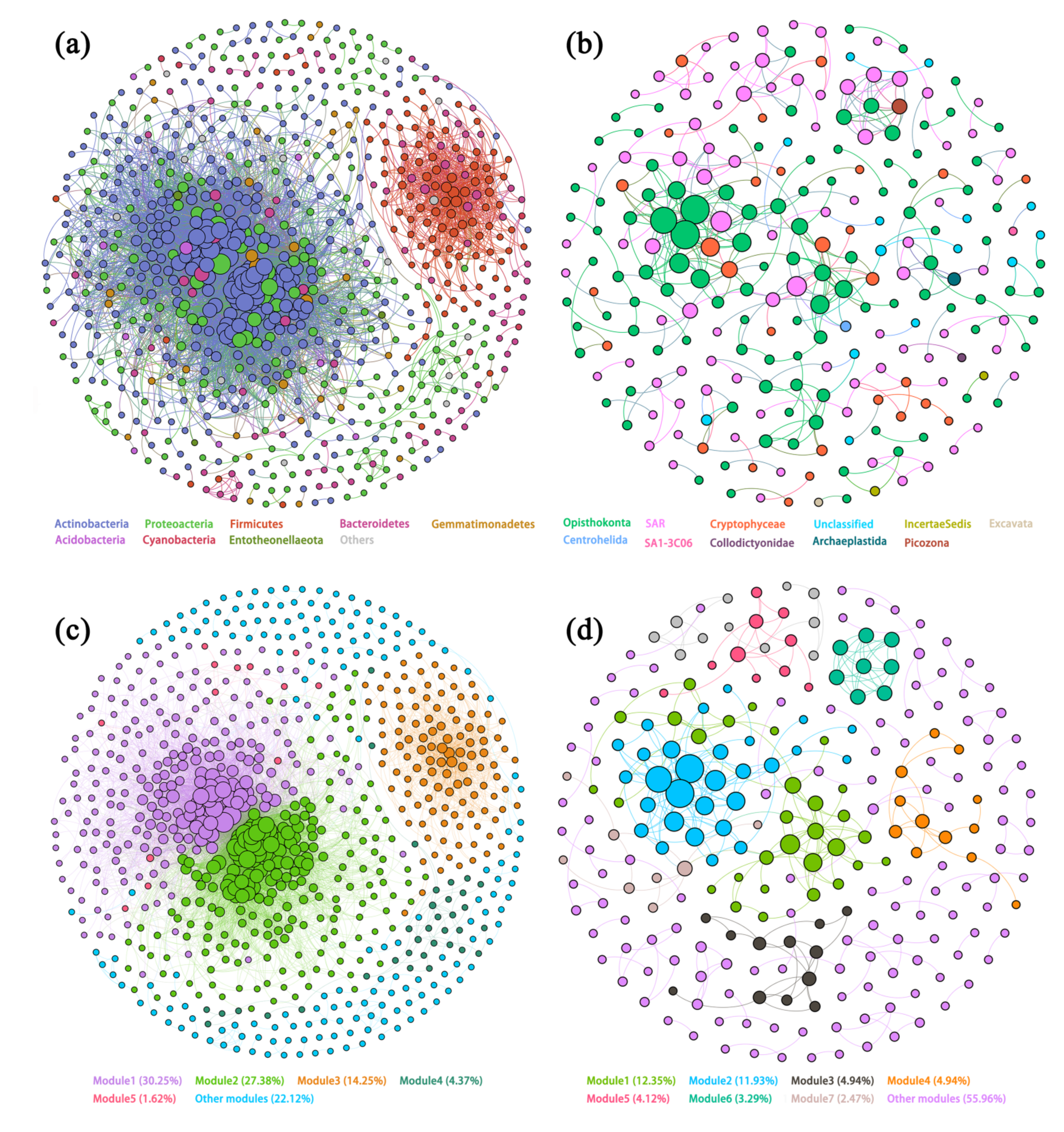
3.4. Co-Occurrence Networks of Different Microbial Communities
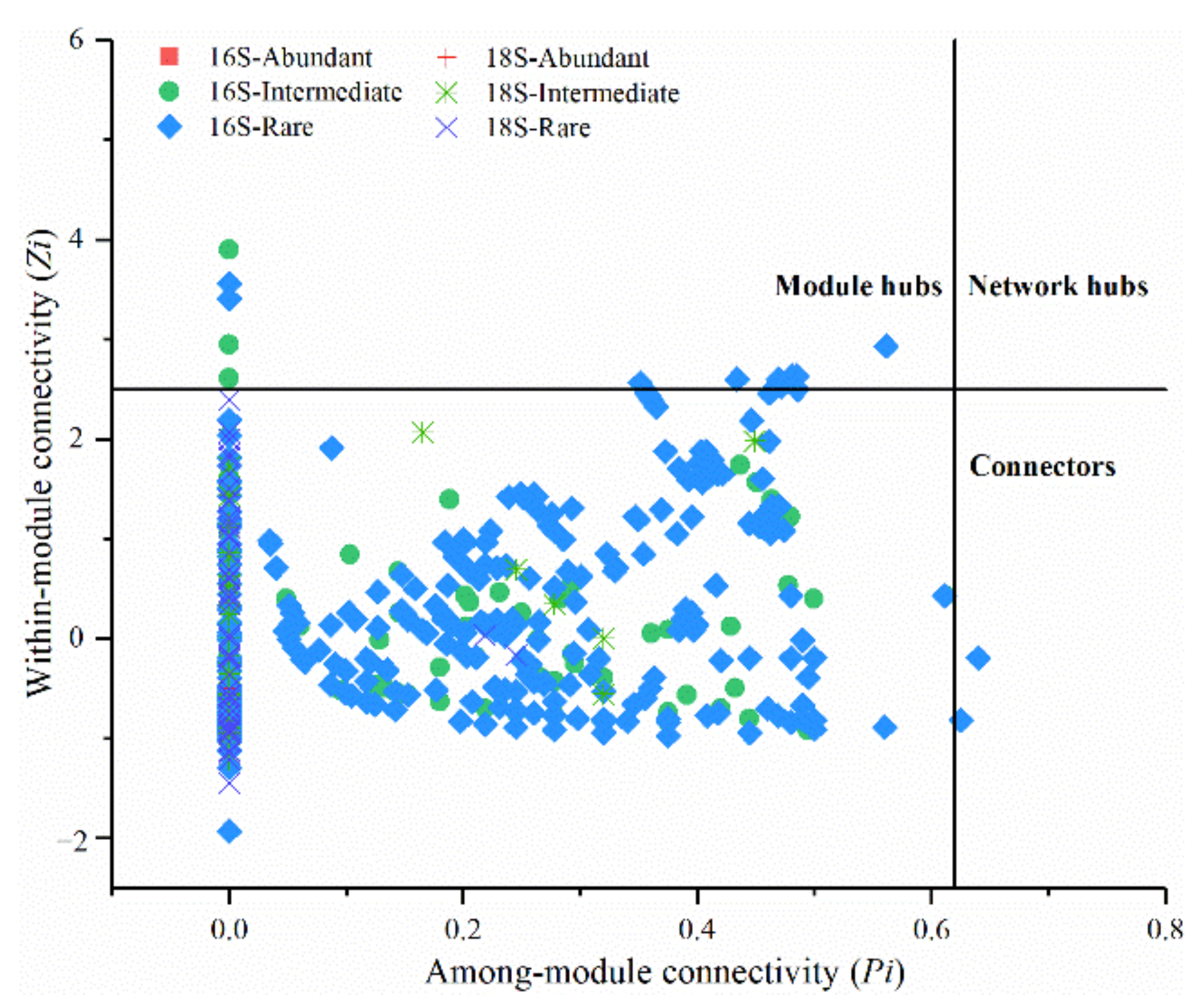
3.5. Modularity and Keystone Species of Networks
4. Discussion
4.1. Patterns of Bacterial and Microeukaryotic Communities
4.2. The Controlling Factors of the Microbial Community Pattern
4.3. Co-Occurrence Networks of Bacterial and Microeukaryotic Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.E.; Cardinale, B.J.; France, K.E.; McIntyre, P.B.; Thebault, E.; Loreau, M. The functional role of biodiversity in ecosystems: Incorporating trophic complexity. Ecol. Lett. 2007, 10, 522–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Kusel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Costello, E.K.; Hamady, M.; Lozupone, C.; Jiang, L.; Schmidt, S.K.; Fierer, N.; Townsend, A.R.; Cleveland, C.C.; Stanish, L.; et al. Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 2011, 13, 135–144. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Chen, J.; Wang, P.; Wang, C.; Wang, X.; Miao, L.; Liu, S.; Yuan, Q.; Sun, S. Distinct Assembly Mechanisms Underlie Similar Biogeographic Patterns of Rare and Abundant Bacterioplankton in Cascade Reservoirs of a Large River. Front. Microbiol. 2020, 11, 158. [Google Scholar] [CrossRef] [Green Version]
- Debroas, D.; Hugoni, M.; Domaizon, I. Evidence for an active rare biosphere within freshwater protists community. Mol. Ecol. 2015, 24, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.D.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Jiao, S.; Lu, Y. Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environ. Microbiol. 2020, 22, 1052–1065. [Google Scholar] [CrossRef]
- Jiao, S.; Xu, Y.; Zhang, J.; Lu, Y. Environmental filtering drives distinct continental atlases of soil archaea between dryland and wetland agricultural ecosystems. Microbiome 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pan, Y.; Yang, J.; Chen, H.; Holohan, B.; Vaudrey, J.; Lin, S.; McManus, G.B. The diversity and biogeography of abundant and rare intertidal marine microeukaryotes explained by environment and dispersal limitation. Environ. Microbiol. 2018, 20, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugoni, M.; Taib, N.; Debroas, D.; Domaizon, I.; Jouan Dufournel, I.; Bronner, G.; Salter, I.; Agogue, H.; Mary, I.; Galand, P.E. Structure of the rare archaeal biosphere and seasonal dynamics of active ecotypes in surface coastal waters. Proc. Natl. Acad. Sci. USA 2013, 110, 6004–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio 2014, 5, e01371-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Gao, H. Ecological networks reveal contrasting patterns of bacterial and fungal communities in glacier-fed streams in Central Asia. PeerJ 2019, 7, e7715. [Google Scholar] [CrossRef] [Green Version]
- Widder, S.; Besemer, K.; Singer, G.A.; Ceola, S.; Bertuzzo, E.; Quince, C.; Sloan, W.T.; Rinaldo, A.; Battin, T.J. Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. USA 2014, 111, 12799–12804. [Google Scholar] [CrossRef] [Green Version]
- Du, S.Q.; Van Rompaey, A.; Shi, P.J.; Wang, J.A. A dual effect of urban expansion on flood risk in the Pearl River Delta (China) revealed by land-use scenarios and direct runoff simulation. Nat. Hazards 2015, 77, 111–128. [Google Scholar] [CrossRef]
- Ou, S.Y.; Yang, Q.S.; Luo, X.X.; Zhu, F.; Luo, K.W.; Yang, H. The influence of runoff and wind on the dispersion patterns of suspended sediment in the Zhujiang (Pearl) River Estuary based on MODIS data. Acta Oceanol. Sin. 2019, 38, 26–35. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, R.; Zhuang, D.C.; Lu, Z.L. Spatial gradient differences of ecosystem services supply and demand in the Pearl River Delta region. J. Clean. Prod. 2021, 279, 123849. [Google Scholar] [CrossRef]
- Campbell, B.J.; Yu, L.; Heidelberg, J.F.; Kirchman, D.L. Activity of abundant and rare bacteria in a coastal ocean. Proc. Natl. Acad. Sci. USA 2011, 108, 12776–12781. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The smallest of all worlds: Pollination networks. J. Theor. Biol. 2006, 240, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Lu, Y. Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Change Biol. 2020, 26, 4506–4520. [Google Scholar] [CrossRef] [PubMed]
- Haro-Moreno, J.M.; Rodriguez-Valera, F.; Rosselli, R.; Martinez-Hernandez, F.; Roda-Garcia, J.J.; Gomez, M.L.; Fornas, O.; Martinez-Garcia, M.; Lopez-Perez, M. Ecogenomics of the SAR11 clade. Environ. Microbiol. 2020, 22, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, S.J. SAR11 Bacteria: The Most Abundant Plankton in the Oceans. Annu. Rev. Mar. Sci. 2017, 9, 231–255. [Google Scholar] [CrossRef]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, F.; Wu, S.; Wu, J.; Yan, J.; Xu, K.; Hong, Y. Biogeographic pattern of bacterioplanktonic community and potential function in the Yangtze River: Roles of abundant and rare taxa. Sci. Total Environ. 2020, 747, 141335. [Google Scholar] [CrossRef]
- Liu, J.; Tu, T.; Gao, G.; Bartlam, M.; Wang, Y. Biogeography and Diversity of Freshwater Bacteria on a River Catchment Scale. Microb. Ecol. 2019, 78, 324–335. [Google Scholar] [CrossRef]
- Porter, K.G. Selective Grazing and Differential Digestion of Algae by Zooplankton. Nature 1973, 244, 179–180. [Google Scholar] [CrossRef]
- Cavalier-Smith, T.; Chao, E.E. Phylogeny and classification of phylum Cercozoa (Protozoa). Protist 2003, 154, 341–358. [Google Scholar] [CrossRef] [Green Version]
- Sherr, E.B.; Sherr, B.F. Significance of predation by protists in aquatic microbial food webs. Antonie Van Leeuwenhoek 2002, 81, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhou, Y.; Yao, X.; Cai, J.; Liu, X.; Tang, X.; Zhang, Y.; Jang, K.S.; Jeppesen, E. Decreasing diversity of rare bacterial subcommunities relates to dissolved organic matter along permafrost thawing gradients. Environ. Int. 2020, 134, 105330. [Google Scholar] [CrossRef]
- Pedros-Alio, C. Marine microbial diversity: Can it be determined? Trends Microbiol. 2006, 14, 257–263. [Google Scholar] [CrossRef]
- Jiao, S.; Wang, J.; Wei, G.; Chen, W.; Lu, Y. Dominant role of abundant rather than rare bacterial taxa in maintaining agro-soil microbiomes under environmental disturbances. Chemosphere 2019, 235, 248–259. [Google Scholar] [CrossRef]
- Jones, A.C.; Hambright, K.D.; Caron, D.A. Ecological Patterns Among Bacteria and Microbial Eukaryotes Derived from Network Analyses in a Low-Salinity Lake. Microb. Ecol. 2018, 75, 917–929. [Google Scholar] [CrossRef]
- De Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, J.J.; van Altena, C.; de Ruiter, P.C.; van Gerven, L.P.A.; Janse, J.H.; Mooij, W.M. Food-web stability signals critical transitions in temperate shallow lakes. Nat. Commun. 2015, 6, 7727. [Google Scholar] [CrossRef] [Green Version]
- De Vries, F.T.; Wallenstein, M.D. Below-ground connections underlying above-ground food production: A framework for optimising ecological connections in the rhizosphere. J. Ecol. 2017, 105, 913–920. [Google Scholar] [CrossRef] [Green Version]
- De Vries, F.T.; Liiri, M.E.; Bjørnlund, L.; Bowker, M.A.; Christensen, S.; Setälä, H.M.; Bardgett, R.D. Land use alters the resistance and resilience of soil food webs to drought. Nat. Clim. Change 2012, 2, 276–280. [Google Scholar] [CrossRef]
- Mikhailov, I.S.; Zakharova, Y.R.; Bukin, Y.S.; Galachyants, Y.P.; Petrova, D.P.; Sakirko, M.V.; Likhoshway, Y.V. Co-occurrence Networks Among Bacteria and Microbial Eukaryotes of Lake Baikal During a Spring Phytoplankton Bloom. Microb. Ecol. 2019, 77, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Q.; Li, X.; Wang, M.; Liu, X.; Yang, J.; Xu, J.; Jiang, Y. Spatial patterns and co-occurrence networks of microbial communities related to environmental heterogeneity in deep-sea surface sediments around Yap Trench, Western Pacific Ocean. Sci. Total Environ. 2021, 759, 143799. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, Y.; Jia, X.H.; Wang, M.M.; Ding, J.J.; Cheng, L.; Bao, F.; Wu, B. Network analysis reveals the strengthening of microbial interaction in biological soil crust development in the Mu Us Sandy Land, northwestern China. Soil Biol. Biochem. 2020, 144, 107782. [Google Scholar] [CrossRef]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Topological Parameters | Bacteria | Microeukaryotes |
|---|---|---|
| Number of edges | 10,360 | 350 |
| Number of nodes | 800 | 243 |
| Network density | 0.0324 | 0.0119 |
| Characteristic degree | 25.9000 | 2.8807 |
| Characteristic path length | 2.8251 | 3.5617 |
| Clustering coefficient | 0.5416 | 0.5343 |
| Modularity | 0.3868 | 0.8538 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Ren, Z.; Ma, J.; Chen, N.; Liu, J. Compositional Changes and Co-Occurrence Patterns of Planktonic Bacteria and Microeukaryotes in a Subtropical Estuarine Ecosystem, the Pearl River Delta. Water 2022, 14, 1227. https://doi.org/10.3390/w14081227
Ma K, Ren Z, Ma J, Chen N, Liu J. Compositional Changes and Co-Occurrence Patterns of Planktonic Bacteria and Microeukaryotes in a Subtropical Estuarine Ecosystem, the Pearl River Delta. Water. 2022; 14(8):1227. https://doi.org/10.3390/w14081227
Chicago/Turabian StyleMa, Kang, Ze Ren, Jiaming Ma, Nannan Chen, and Jingling Liu. 2022. "Compositional Changes and Co-Occurrence Patterns of Planktonic Bacteria and Microeukaryotes in a Subtropical Estuarine Ecosystem, the Pearl River Delta" Water 14, no. 8: 1227. https://doi.org/10.3390/w14081227
APA StyleMa, K., Ren, Z., Ma, J., Chen, N., & Liu, J. (2022). Compositional Changes and Co-Occurrence Patterns of Planktonic Bacteria and Microeukaryotes in a Subtropical Estuarine Ecosystem, the Pearl River Delta. Water, 14(8), 1227. https://doi.org/10.3390/w14081227