
Wastewater Microbiome Analysis for Population Alcohol Abuse

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Wastewater Sampling
2.2. DNA Extraction and Nanopore Sequencing
2.3. Metagenomic Data Analysis
2.4. Microbiome Analysis for Biomarkers of Alcoholism
3. Results and Discussion
3.1. Microbial Communities in Wastewater Samples
3.2. Human Gut Microbiomes Detected in Wastewater
3.3. Impacts of Human Health Risk Factors on Human Gut Microbiome in Wastewater
3.4. Potential Microbial Biomarkers of Alcoholism with Smoking and Obesity as Confounding Factors
3.5. Microbiological Function and Metabolism in Relation to Alcohol Consumption
3.6. Implications and Limitations
4. Conclusions
- Environmental microorganisms constitute a significant portion of the sewage microbiota, with their composition primarily shaped by geographic locations.
- Major human health risk factors including alcoholism, smoking, and obesity collectively shape the human gut microbiota in wastewater and exhibit correlations between obesity, smoking and alcoholism.
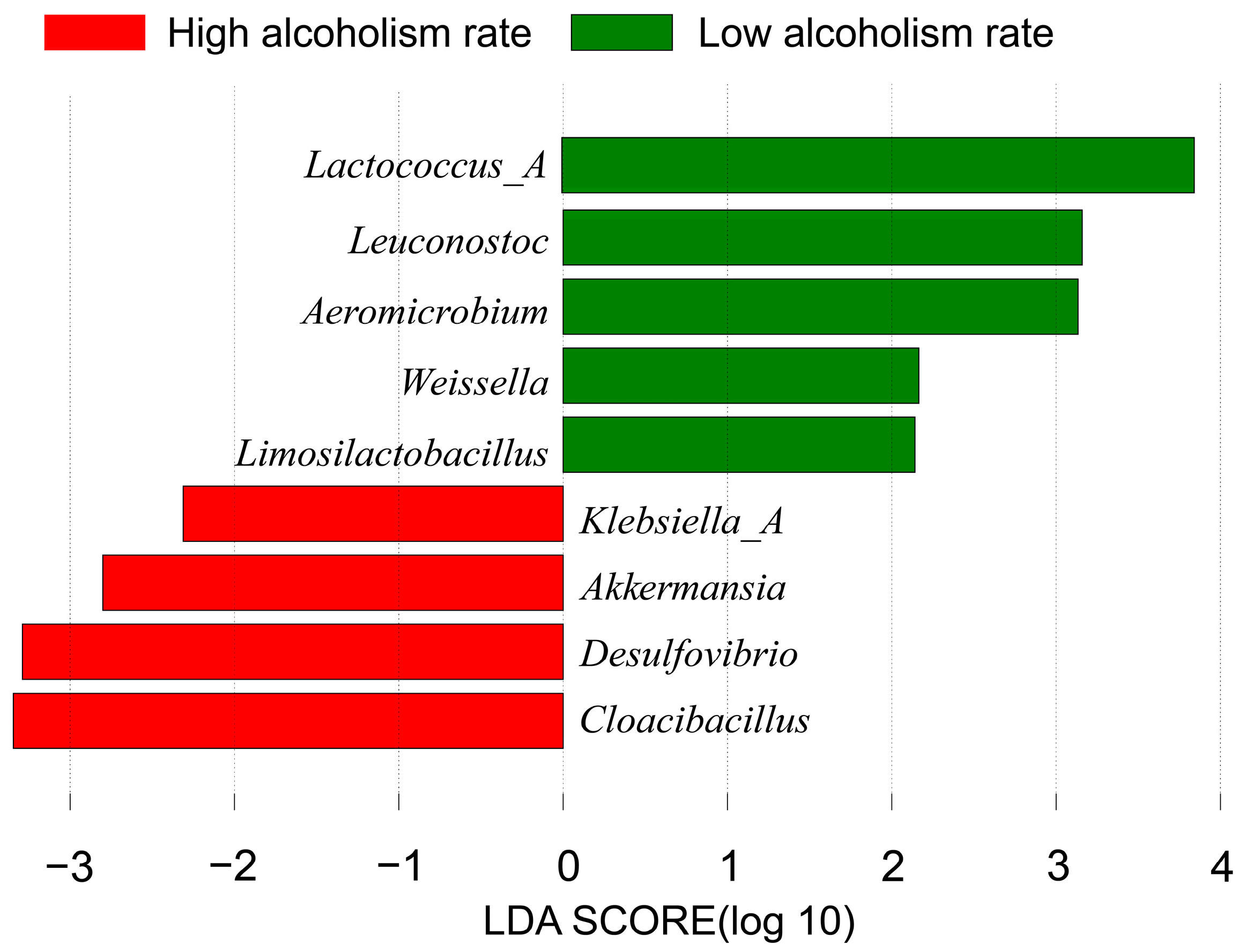
- Several potential biomarkers for alcoholism have been identified using LEfSe, when accounting for confounding factors such as obesity and smoking. These biomarkers include Lactococcus_A, Leuconostoc, Aeromicrobium, Akkermansia, Weissella, Limosilactobacillus, Klebsiella_A, Desulfovibrio and Cloacibacillus. Microbial biomarkers for alcoholism primarily comprise lactic acid bacteria, which produce lactic acid and influence alcohol metabolism.
- Function annotations of the wastewater metagenomes indicate that alcoholism has the potential to enhance host energy metabolism and intercellular signaling while simultaneously compromising the overall immune function in the host organism.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swahn, M.H.; Robow, Z.; Umenze, F.; Balenger, A.; Dumbili, E.W.; Obot, I. A readiness assessment for the prevention of alcohol-related harm in West Africa: A new methodological approach to inform practice and policy. Int. J. Drug Policy 2022, 103, 103650. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, C.; Möckl, J.; Seitz, N.-N.; Wilms, N.; Olderbak, S.; Kraus, L. The Use of Psychoactive Substances in Germany: Findings from the Epidemiological Survey of Substance Abuse 2021. Dtsch. Ärzteblatt Int. 2022, 119, 527. [Google Scholar]
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans—From biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, J.-E. Impact of drinking alcohol on gut microbiota: Recent perspectives on ethanol and alcoholic beverage. Curr. Opin. Food Sci. 2021, 37, 91–97. [Google Scholar] [CrossRef]
- Martino, C.; Zaramela, L.S.; Gao, B.; Embree, M.; Tarasova, J.; Parker, S.J.; Wang, Y.; Chu, H.; Chen, P.; Lee, K.-C. Acetate reprograms gut microbiota during alcohol consumption. Nat. Commun. 2022, 13, 4630. [Google Scholar] [CrossRef] [PubMed]
- Thomes, P.G.; Rasineni, K.; Saraswathi, V.; Kharbanda, K.K.; Clemens, D.L.; Sweeney, S.A.; Kubik, J.L.; Donohue, T.M., Jr.; Casey, C.A. Natural recovery by the liver and other organs after chronic alcohol use. Alcohol Res. Curr. Rev. 2021, 41, 5. [Google Scholar] [CrossRef]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 2020, 11, 5206. [Google Scholar] [CrossRef]
- Rosas-Plaza, S.; Hernández-Terán, A.; Navarro-Díaz, M.; Escalante, A.E.; Morales-Espinosa, R.; Cerritos, R. Human gut microbiome across different lifestyles: From hunter-gatherers to urban populations. Front. Microbiol. 2022, 13, 843170. [Google Scholar] [CrossRef]
- Ojima, M.; Motooka, D.; Shimizu, K.; Gotoh, K.; Shintani, A.; Yoshiya, K.; Nakamura, S.; Ogura, H.; Iida, T.; Shimazu, T. Metagenomic analysis reveals dynamic changes of whole gut microbiota in the acute phase of intensive care unit patients. Dig. Dis. Sci. 2016, 61, 1628–1634. [Google Scholar] [CrossRef]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome. MSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef]
- Li, F.; Yang, S.; Zhang, L.; Qiao, L.; Wang, L.; He, S.; Li, J.; Yang, N.; Yue, B.; Zhou, C. Comparative metagenomics analysis reveals how the diet shapes the gut microbiota in several small mammals. Ecol. Evol. 2022, 12, e8470. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Pires, J.; Silvester, R.; Zhao, C.; Song, J.; Criscuolo, N.G.; Gilbert, M.; Bonhoeffer, S.; Laxminarayan, R. Global trends in antimicrobial resistance in animals in low-and middle-income countries. Science 2019, 365, eaaw1944. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Kjellerup, B.V. Predicting the potential for organohalide respiration in wastewater: Comparison of intestinal and wastewater microbiomes. Sci. Total Environ. 2020, 705, 135833. [Google Scholar] [CrossRef]
- Guo, B.; Liu, C.; Gibson, C.; Frigon, D. Wastewater microbial community structure and functional traits change over short timescales. Sci. Total Environ. 2019, 662, 779–785. [Google Scholar] [CrossRef]
- García-Aljaro, C.; Blanch, A.R.; Campos, C.; Jofre, J.; Lucena, F. Pathogens, faecal indicators and human-specific microbial source-tracking markers in sewage. J. Appl. Microbiol. 2019, 126, 701–717. [Google Scholar] [CrossRef]
- McLellan, S.L.; Roguet, A. The unexpected habitat in sewer pipes for the propagation of microbial communities and their imprint on urban waters. Curr. Opin. Biotechnol. 2019, 57, 34–41. [Google Scholar] [CrossRef]
- Nimonkar, Y.S.; Yadav, B.; Talreja, P.; Sharma, A.; Patil, S.; Saware, S.S.; Ranade, D.R.; Prakash, O. Assessment of the role of wastewater treatment plant in spread of antibiotic resistance and bacterial pathogens. Indian J. Microbiol. 2019, 59, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Eun, J.W.; Cho, H.J.; Song, D.S.; Kim, C.W.; Kim, Y.S.; Lee, S.W.; Kim, Y.K.; Yang, J.; Choi, J. Microbiome as a potential diagnostic and predictive biomarker in severe alcoholic hepatitis. Aliment. Pharmacol. Ther. 2021, 53, 540–551. [Google Scholar] [CrossRef]
- Zafari, N.; Velayati, M.; Fahim, M.; Maftouh, M.; Pourali, G.; Khazaei, M.; Nassiri, M.; Hassanian, S.M.; Ghayour-Mobarhan, M.; Ferns, G.A. Role of gut bacterial and non-bacterial microbiota in alcohol-associated liver disease: Molecular mechanisms, biomarkers, and therapeutic prospective. Life Sci. 2022, 305, 120760. [Google Scholar] [CrossRef]
- Chen, J.; Ryu, E.; Hathcock, M.; Ballman, K.; Chia, N.; Olson, J.E.; Nelson, H. Impact of demographics on human gut microbial diversity in a US Midwest population. PeerJ 2016, 4, e1514. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, S.; Chen, Y.; Zhao, J.; Prosun, T.; O’Brien, J.W.; Mueller, J.F.; Tscharke, B.J.; Coin, L.J.M.; Luby, S.P. Associations between Wastewater Microbiome and Population Smoking Rate Identified Using Wastewater-Based Epidemiology. Environ. Health 2023, 1, 394–404. [Google Scholar] [CrossRef]
- Australian Bureau of Statistics. National Health Survey: First Results 2017–2018; ABS: Canberra, Australia, 2018.
- O‘Brien, J.; Grant, S.; Mueller, J.; Tscharke, B.; Gerber, C.; White, J. National Wastewater Drug Monitoring Program: Report 1; Australian Criminal Intelligence Commission: Canberra, Australia, 2017.
- O‘Brien, J.W.; Grant, S.; Banks, A.P.W.; Bruno, R.; Carter, S.; Choi, P.M.; Covaci, A.; Crosbie, N.D.; Gartner, C.; Hall, W.; et al. A National Wastewater Monitoring Program for a better understanding of public health: A case study using the Australian Census. Environ. Int. 2018, 122, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Kingsford, C. Salmon: Accurate, versatile and ultrafast quantification from RNA-seq data using lightweight-alignment. BioRxiv 2015. [Google Scholar] [CrossRef]
- Safder, I.; Shao, G.; Sheng, Z.; Hu, P.; Tang, S. Genome-wide identification studies–A primer to explore new genes in plant species. Plant Biol. 2022, 24, 9–22. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Newton, R.J.; McLellan, S.L.; Dila, D.K.; Vineis, J.H.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. Sewage reflects the microbiomes of human populations. MBio 2015, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36, D250–D254. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O‘Hara, R.B.; Simpson, G.L.; Solymos, P. Package ‘vegan’. Community Ecol. Package Version 2019, 2, 1–295. [Google Scholar]
- Abdugheni, R.; Wang, W.Z.; Wang, Y.J.; Du, M.X.; Liu, F.L.; Zhou, N.; Jiang, C.Y.; Wang, C.Y.; Wu, L.; Ma, J. Metabolite profiling of human-originated Lachnospiraceae at the strain level. iMeta 2022, 1, e58. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, M. Gephi: Introduction to Network Analysis and Visualization; University of Lausanne: Lausanne, Switzerland, 2015. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut microbial community structure and complications following kidney transplantation: A pilot study. Transplantation 2014, 98, 697. [Google Scholar]
- Shanks, O.C.; Newton, R.J.; Kelty, C.A.; Huse, S.M.; Sogin, M.L.; McLellan, S.L. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl. Environ. Microbiol. 2013, 79, 2906–2913. [Google Scholar] [CrossRef]
- Zhao, C.; Shi, Z.J.; Pollard, K.S. Pitfalls of genotyping microbial communities with rapidly growing genome collections. Cell Syst. 2023, 14, 160–176.e163. [Google Scholar] [CrossRef]
- Su, J.-Q.; An, X.-L.; Li, B.; Chen, Q.-L.; Gillings, M.R.; Chen, H.; Zhang, T.; Zhu, Y.-G. Metagenomics of urban sewage identifies an extensively shared antibiotic resistome in China. Microbiome 2017, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.R.; Dey, J.; Jaiswal, A.; Roy, R.; Misra, N.; Suar, M. Immunoinformatics-guided designing of epitope-based subunit vaccine from Pilus assembly protein of Acinetobacter baumannii bacteria. J. Immunol. Methods 2022, 508, 113325. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Hirai, H.; Che, F.-S.; Choi, S. Molecular Basis for the Activation of Human Innate Immune Response by the Flagellin Derived from Plant-Pathogenic Bacterium, Acidovorax avenae. Int. J. Mol. Sci. 2021, 22, 6920. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Ju, F.; Zhang, T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol. 2014, 98, 3317–3326. [Google Scholar] [CrossRef]
- Scapellato, G. Cloacibacterium: Developing a Model System to Study the Bacteria of Wastewater Conveyance Systems. In Proceedings of the UWM Undergraduate Research Symposium, Milwaukee, WI, USA, 10 May 2022. [Google Scholar]
- Venâncio, I.; Luís, Â.; Domingues, F.; Oleastro, M.; Pereira, L.; Ferreira, S. The prevalence of Arcobacteraceae in aquatic environments: A systematic review and meta-analysis. Pathogens 2022, 11, 244. [Google Scholar] [CrossRef]
- van Den Brule, S.; Ambroise, J.; Lecloux, H.; Levard, C.; Soulas, R.; De Temmerman, P.-J.; Palmai-Pallag, M.; Marbaix, E.; Lison, D. Dietary silver nanoparticles can disturb the gut microbiota in mice. Part. Fibre Toxicol. 2015, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, R.S.; Munk, P.; Njage, P.; Van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- LaMartina, E.L.; Mohaimani, A.A.; Newton, R.J. Urban wastewater bacterial communities assemble into seasonal steady states. Microbiome 2021, 9, 116. [Google Scholar] [CrossRef]
- Ramos-Sevillano, E.; Ercoli, G.; Brown, J.S. Mechanisms of naturally acquired immunity to Streptococcus pneumoniae. Front. Immunol. 2019, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Liu, Y.; Chen, X.; He, J.; Yu, Y.; Wang, Y.; Bai, J. Dynamic changes and effects of environmental tobacco smoke exposure and weaning time on gut microbiota in young children aged 0–2 years. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Ozato, N.; Saito, S.; Yamaguchi, T.; Katashima, M.; Tokuda, I.; Sawada, K.; Katsuragi, Y.; Kakuta, M.; Imoto, S.; Ihara, K.; et al. Blautia genus associated with visceral fat accumulation in adults 20–76 years of age. npj Biofilms Microbiomes 2019, 5, 28. [Google Scholar] [CrossRef]
- Gryaznova, M.; Smirnova, Y.; Burakova, I.; Syromyatnikov, M.; Chizhkov, P.; Popov, E.; Popov, V. Changes in the Human Gut Microbiome Caused by the Short-Term Impact of Lactic Acid Bacteria Consumption in Healthy People. Probiotics Antimicrob. Proteins 2023. [Google Scholar] [CrossRef]
- Martinez, E.; Crevecoeur, S.; Thirion, C.; Grandjean, J.; Fall, P.A.; Hayette, M.-P.; Michel, M.; Taminiau, B.; Louis, E.; Daube, G. Gut Microbiota Associated with Clostridioides difficile Carriage in Three Clinical Groups (Inflammatory Bowel Disease, C. difficile Infection and Healthcare Workers) in Hospital Field. Microorganisms 2023, 11, 2527. [Google Scholar] [CrossRef] [PubMed]
- Coureuil, M.; Jamet, A.; Bille, E.; Lécuyer, H.; Bourdoulous, S.; Nassif, X. Molecular interactions between Neisseria meningitidis and its human host. Cell. Microbiol. 2019, 21, e13063. [Google Scholar] [CrossRef]
- Młynarczyk-Bonikowska, B.; Majewska, A.; Malejczyk, M.; Młynarczyk, G.; Majewski, S. Multiresistant Neisseria gonorrhoeae: A new threat in second decade of the XXI century. Med. Microbiol. Immunol. 2020, 209, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Walters, S.P.; Field, K.G. Survival and persistence of human and ruminant-specific faecal Bacteroidales in freshwater microcosms. Environ. Microbiol. 2009, 11, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Q.; Peng, C.; Ruan, B. Both gut microbiota and differentially expressed proteins are relevant to the development of obesity. BioMed Res. Int. 2020, 2020, 5376108. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.; Zhao, J.; Prosun, T.; O‘Brien, J.W.; Coin, L.; Hai, F.I.; Sanderson-Smith, M.; Bi, P.; Jiang, G. Associations between the wastewater microbiome and community obesity rates: A study in Australia. Soil Environ. Health 2024, 2, 100081. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.; Rodriguez-Palacios, A. The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Majumder, R.; Debnath, A.; Das, A. Potential role of healthy microbiome in metabolic syndrome and immune competence. In Viral, Parasitic, Bacterial, and Fungal Infections; Elsevier: Amsterdam, The Netherlands, 2023; pp. 805–814. [Google Scholar]
- Zhang, J.; Zhang, F.; Zhao, C.; Xu, Q.; Liang, C.; Yang, Y.; Wang, H.; Shang, Y.; Wang, Y.; Mu, X. Dysbiosis of the gut microbiome is associated with thyroid cancer and thyroid nodules and correlated with clinical index of thyroid function. Endocrine 2019, 64, 564–574. [Google Scholar] [CrossRef]
- Hiippala, K.; Barreto, G.; Burrello, C.; Diaz-Basabe, A.; Suutarinen, M.; Kainulainen, V.; Bowers, J.R.; Lemmer, D.; Engelthaler, D.M.; Eklund, K.K. Novel Odoribacter splanchnicus strain and its outer membrane vesicles exert immunoregulatory effects in vitro. Front. Microbiol. 2020, 11, 575455. [Google Scholar] [CrossRef]
- Plaza-Díaz, J.; Manzano, M.; Ruiz-Ojeda, F.J.; Giron, M.D.; Salto, R.; López-Pedrosa, J.M.; Santos-Fandila, A.; Garcia-Corcoles, M.T.; Rueda, R.; Gil, Á. Intake of slow-digesting carbohydrates is related to changes in the microbiome and its functional pathways in growing rats with obesity induced by diet. Front. Nutr. 2022, 9, 992682. [Google Scholar] [CrossRef]
- Kosnicki, K.L.; Penprase, J.C.; Cintora, P.; Torres, P.J.; Harris, G.L.; Brasser, S.M.; Kelley, S.T. Effects of moderate, voluntary ethanol consumption on the rat and human gut microbiome. Addict. Biol. 2019, 24, 617–630. [Google Scholar] [CrossRef]
- Hampson, D.J. The spirochete Brachyspira pilosicoli, enteric pathogen of animals and humans. Clin. Microbiol. Rev. 2018, 31, e00087-17. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, L.; Wang, X.; Yi, Y.; Shan, Y.; Liu, B.; Zhou, Y.; Lü, X. Roles of intestinal Parabacteroides in human health and diseases. FEMS Microbiol. Lett. 2022, 369, fnac072. [Google Scholar] [CrossRef] [PubMed]
- Huber-Ruano, I.; Calvo, E.; Mayneris-Perxachs, J.; Rodríguez-Peña, M.M.; Ceperuelo-Mallafré, V.; Cedó, L.; Núñez-Roa, C.; Miro-Blanch, J.; Arnoriaga-Rodríguez, M.; Balvay, A. Orally administered Odoribacter laneus improves glucose control and inflammatory profile in obese mice by depleting circulating succinate. Microbiome 2022, 10, 135. [Google Scholar] [CrossRef]
- Lemmer, P.; Manka, P.; Best, J.; Kahraman, A.; Kälsch, J.; Vilchez-Vargas, R.; Link, A.; Chiang, H.; Gerken, G.; Canbay, A. Effects of moderate alcohol consumption in non-alcoholic fatty liver disease. J. Clin. Med. 2022, 11, 890. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Chu, C.W.; Khoo, J.J.C.; Cheong, M.; Soon, G.H.; Ho, E.X.P.; Law, N.M.; De Sessions, P.F.; Fock, K.M.; Ang, T.L. A pilot study to examine the association between human gut microbiota and the host’s central obesity. JGH Open 2019, 3, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Xu, M.; Xing, Y.; Chen, C.; Jiang, J.; Xu, X. Effects of Cigarette Smoke Exposure on the Gut Microbiota and Liver Transcriptome in Mice Reveal Gut–Liver Interactions. Int. J. Mol. Sci. 2022, 23, 11008. [Google Scholar] [CrossRef]
- Abdul-Mutalib, N.-A.; Nordin, S.A.; Osman, M.; Roslan, A.M.; Ishida, N.; Sakai, K.; Tashiro, Y.; Tashiro, K.; Maeda, T.; Shirai, Y. The prevalence of foodborne pathogenic bacteria on cutting boards and their ecological correlation with background biota. AIMS Microbiol. 2016, 2, 138–151. [Google Scholar] [CrossRef]
- Khelaifia, S.; Raoult, D. Haloferax massiliensis sp. nov., the first human-associated halophilic archaea. New Microbes New Infect. 2016, 12, 96. [Google Scholar] [CrossRef]
- Shao, M.; Zhang, C.; Wang, X.; Wang, N.; Chen, Q.; Cui, G.; Xu, Q. Co-digestion of food waste and hydrothermal liquid digestate: Promotion effect of self-generated hydrochars. Environ. Sci. Ecotechnology 2023, 15, 100239. [Google Scholar] [CrossRef]
- Huo, R.-X.; Wang, Y.-J.; Hou, S.-B.; Wang, W.; Zhang, C.-Z.; Wan, X.-H. Gut mucosal microbiota profiles linked to colorectal cancer recurrence. World J. Gastroenterol. 2022, 28, 1946. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Neylon, E.; Nyhan, L.; Zannini, E.; Monin, T.; Münch, S.; Sahin, A.W.; Arendt, E.K. Food Ingredients for the Future: In-Depth Analysis of the Effects of Lactic Acid Bacteria Fermentation on Spent Barley Rootlets. Fermentation 2023, 9, 78. [Google Scholar] [CrossRef]
- Konkit, M.; Choi, W.J.; Kim, W. Aldehyde dehydrogenase activity in Lactococcus chungangensis: Application in cream cheese to reduce aldehyde in alcohol metabolism. J. Dairy Sci. 2016, 99, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Shimizu, H.; Hisa, K.; Matsuzaki, C.; Inuki, S.; Ando, Y.; Nishida, A.; Izumi, A.; Yamano, M.; Ushiroda, C. Host metabolic benefits of prebiotic exopolysaccharides produced by Leuconostoc mesenteroides. Gut Microbes 2023, 15, 2161271. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Park, J.Y.; Jeong, H.R.; Heo, H.J.; Han, N.S.; Kim, J.H. Probiotic properties of Weissella strains isolated from human faeces. Anaerobe 2012, 18, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.G.; Fusieger, A.; Milião, G.L.; Martins, E.; Drider, D.; Nero, L.A.; de Carvalho, A.F. Weissella: An emerging bacterium with promising health benefits. Probiotics Antimicrob. Proteins 2021, 13, 915–925. [Google Scholar] [CrossRef]
- Saviano, A.; Mattia, B.; Migneco, A.; Gunawardena, G.; Zanza, C.; Candelli, M.; Franceschi, F.; Ojetti, V. Lactobacillus Reuteri DSM 17938 (Limosilactobacillus reuteri) in Diarrhea and Constipation: Two Sides of the Same Coin? Medicina 2021, 57, 643. [Google Scholar] [CrossRef]
- Shellito, J.E.; Zheng, M.Q.; Ye, P.; Ruan, S.; Shean, M.K.; Kolls, J. Effect of alcohol consumption on host release of interleukin-17 during pulmonary infection with Klebsiella pneumoniae. Alcohol. Clin. Exp. Res. 2001, 25, 872–881. [Google Scholar]
- Guo, D.; Park, C.; Li, Y.; Li, B.; Yang, Q.; Deng, Y.; Gao, N.L.; Li, R.; Wang, X.; Yi, L. Akkermansia muciniphila ameliorates depressive disorders in a murine alcohol-LPS (mALPS) model. Food Funct. 2022, 13, 12766–12776. [Google Scholar] [CrossRef]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zhou, L.Z.; Fang, S.T.; Long, H.Y.; Chen, J.Y.; Zhang, G.X. Isolation of Desulfovibrio spp. from human gut microbiota using a next-generation sequencing directed culture method. Lett. Appl. Microbiol. 2019, 68, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Domingo, M.-C.; Yansouni, C.; Gaudreau, C.; Lamothe, F.; Lévesque, S.; Tremblay, C.; Garceau, R. Cloacibacillus sp., a potential human pathogen associated with bacteremia in Quebec and New Brunswick. J. Clin. Microbiol. 2015, 53, 3380–3383. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Parmeggiani, C.; Faggi, E.; Bonaccini, C.; Gratteri, P.; Sim, L.; Gloster, T.M.; Roberts, S.; Davies, G.J.; Rose, D.R. Total syntheses of casuarine and its 6-O-α-glucoside: Complementary inhibition towards glycoside hydrolases of the GH31 and GH37 families. Chem.–A Eur. J. 2009, 15, 1627–1636. [Google Scholar] [CrossRef]
- Rose, D.R. Structure, mechanism and inhibition of Golgi α-mannosidase II. Curr. Opin. Struct. Biol. 2012, 22, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.; Shekarriz, S.; Surette, M.G. Human gut metagenomes encode diverse GH156 Sialidases. Appl. Environ. Microbiol. 2022, 88, e01755-22. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Chen, Y.-C.; Chen, S.-J.; Lee, C.-H.; Cheng, C.-M. Alcohol addiction, gut microbiota, and alcoholism treatment: A review. Int. J. Mol. Sci. 2020, 21, 6413. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazmi, H.E.; Mohammadi, A.; Hejna, A.; Majtacz, J.; Esmaeili, A.; Habibzadeh, S.; Saeb, M.R.; Badawi, M.; Lima, E.C.; Mąkinia, J. Wastewater treatment for reuse in agriculture: Prospects and challenges. Environ. Res. 2023, 236, 116711. [Google Scholar] [CrossRef]
- Paull, P.; Haber, P.S.; Chitty, K.; Seth, D. Evaluation of a novel method for the analysis of alcohol biomarkers: Ethyl glucuronide, ethyl sulfate and phosphatidylethanol. Alcohol 2018, 67, 7–13. [Google Scholar]
- Wang, R.; Gamon, J.A.; Cavender-Bares, J.; Townsend, P.A.; Zygielbaum, A.I. The spatial sensitivity of the spectral diversity–biodiversity relationship: An experimental test in a prairie grassland. Ecol. Appl. 2018, 28, 541–556. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Chen, Y.; Zhao, J.; Prosun, T.A.; O’Brien, J.W.; Coin, L.; Hai, F.I.; Sanderson-Smith, M.; Jiang, G. Wastewater Microbiome Analysis for Population Alcohol Abuse. Water 2024, 16, 2149. https://doi.org/10.3390/w16152149
Wu J, Chen Y, Zhao J, Prosun TA, O’Brien JW, Coin L, Hai FI, Sanderson-Smith M, Jiang G. Wastewater Microbiome Analysis for Population Alcohol Abuse. Water. 2024; 16(15):2149. https://doi.org/10.3390/w16152149
Chicago/Turabian StyleWu, Jiangping, Yan Chen, Jiawei Zhao, Tanjila Alam Prosun, Jake William O’Brien, Lachlan Coin, Faisal I. Hai, Martina Sanderson-Smith, and Guangming Jiang. 2024. "Wastewater Microbiome Analysis for Population Alcohol Abuse" Water 16, no. 15: 2149. https://doi.org/10.3390/w16152149