


Network of Nitrifying Bacteria in Aquarium Biofilters: An Unfaltering Cooperation Between Comammox Nitrospira and Ammonia-Oxidizing Archaea


Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of the Fish Farming Tank and Filtration System
2.2. Sampling and DNA Extracion
2.3. Concentration of Nitrogen Compounds and Dissolved Oxygen
2.4. Metagenomic Sequencing and Data Analysis
2.5. Construction of Microbial Co-Occurrence Network
3. Results
3.1. Results of Physicochemical Measurements
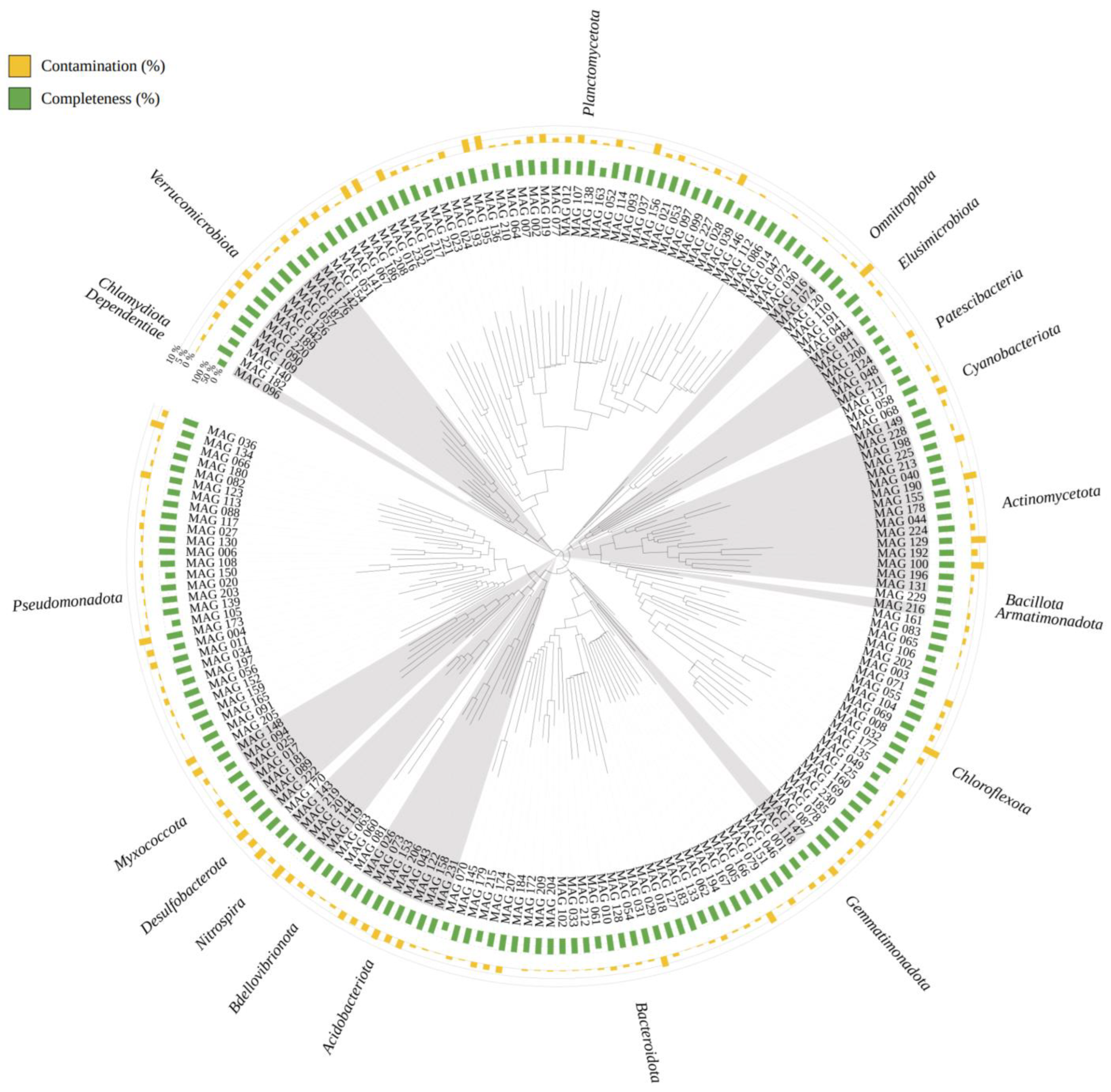
3.2. Genome Reconstruction and Biofilter Microbiome Composition
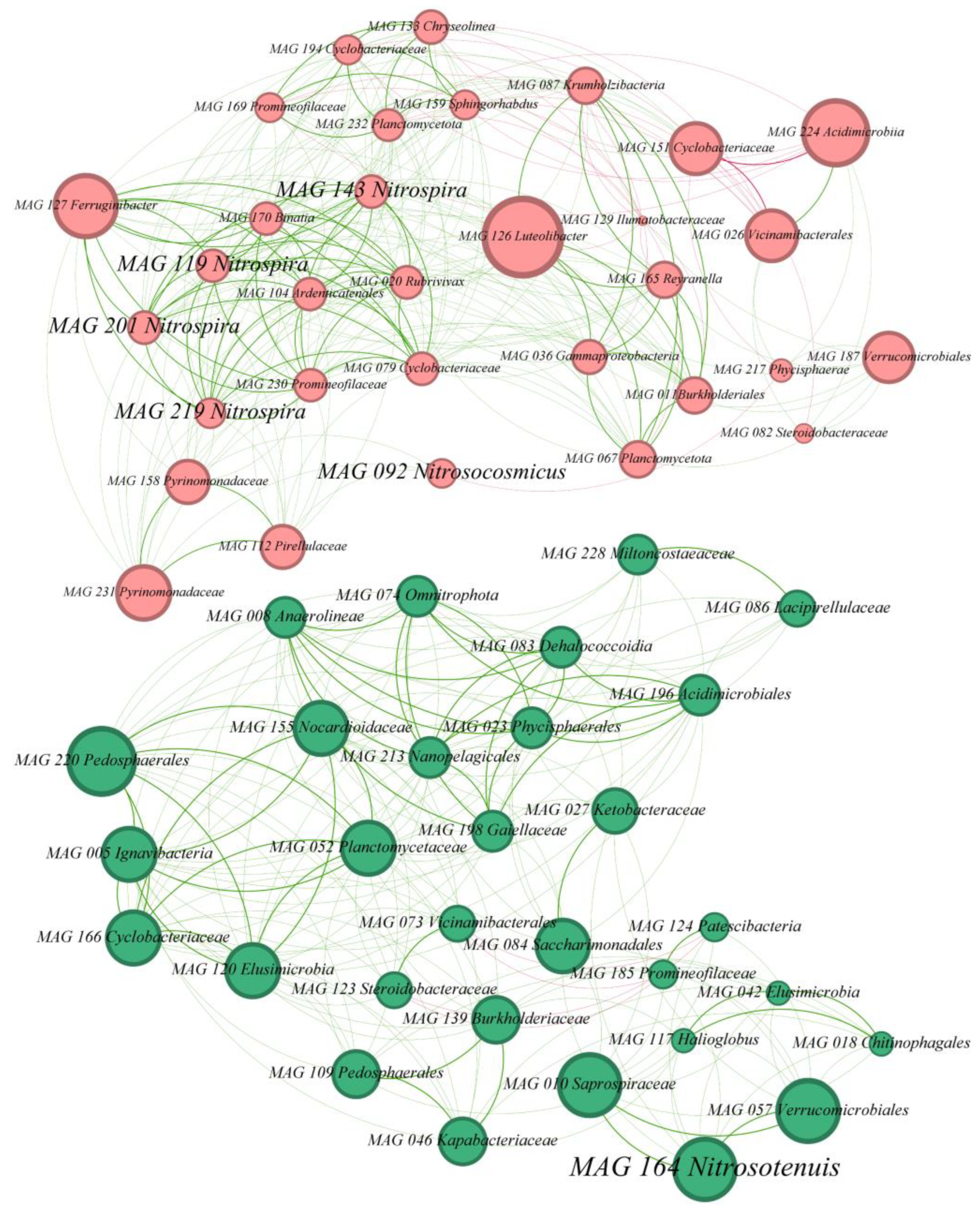
3.3. Biofilter Microbial Network
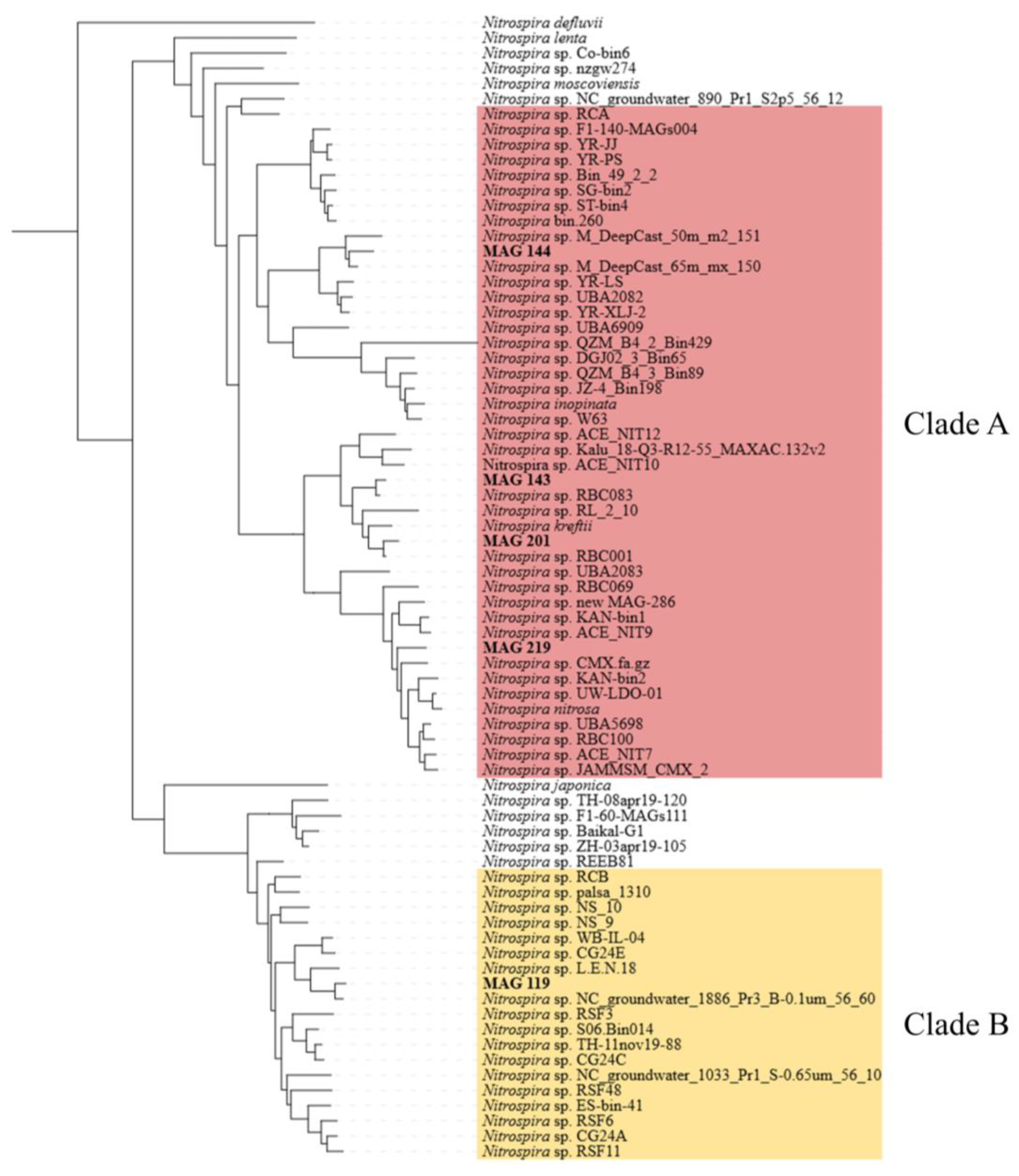
3.4. Nitrogen-Cycle-Related Genes
4. Discussion
4.1. The Diversity and Role of Nitrifiers in the Biofilter
4.2. Diversity and Role of Heterotrophic Bacteria in the Biofilter
4.3. Competitive and Symbiotic Relationships Within the Bacterial Network of the Biofilter
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2024—Blue Transformation in Action; FAO: Rome, Italy, 2024; ISBN 978-92-5-138763-4. [Google Scholar]
- Nyakeya, K.; Masese, F.O.; Gichana, Z.; Nyamora, J.M.; Getabu, A.; Onchieku, J.; Odoli, C.; Nyakwama, R. Cage Farming In The Environmental Mix Of Lake Victoria: An Analysis Of Its Status, Potential Environmental And Ecological Effects, And A Call For Sustainability. Aquat. Ecosyst. Health Manag. 2022, 25, 37–52. [Google Scholar] [CrossRef]
- Thomas, M.; Pasquet, A.; Aubin, J.; Nahon, S.; Lecocq, T. When More Is More: Taking Advantage of Species Diversity to Move towards Sustainable Aquaculture. Biol. Rev. Camb. Philos. Soc. 2021, 96, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.L.; Chin, J.Y.; Mohd Harun, M.H.Z.; Low, S.C. Environmental Impacts and Imperative Technologies towards Sustainable Treatment of Aquaculture Wastewater: A Review. J. Water Process Eng. 2022, 46, 102553. [Google Scholar] [CrossRef]
- Yang, J.; Jia, L.; Guo, Z.; Shen, Y.; Li, X.; Mou, Z.; Yu, K.; Lin, J.C.-W. Prediction and Control of Water Quality in Recirculating Aquaculture System Based on Hybrid Neural Network. Eng. Appl. Artif. Intell. 2023, 121, 106002. [Google Scholar] [CrossRef]
- Jamal, A.; Nasser, A.; van Rijn, J. Real-Time Ammonia Estimation in Recirculating Aquaculture Systems: A Data Assimilation Approach. Aquac. Eng. 2024, 106, 102432. [Google Scholar] [CrossRef]
- Lin, W.; Luo, H.; Wu, J.; Hung, T.-C.; Cao, B.; Liu, X.; Yang, J.; Yang, P. A Review of the Emerging Risks of Acute Ammonia Nitrogen Toxicity to Aquatic Decapod Crustaceans. Water 2022, 15, 27. [Google Scholar] [CrossRef]
- Vaage, B.; Myrick, C. The Effects of Acute and Chronic Exposure of Ammonia on Juvenile Burbot (Lota lota) Growth and Survival. Aquaculture 2021, 542, 736891. [Google Scholar] [CrossRef]
- Zhang, T.-X.; Li, M.-R.; Liu, C.; Wang, S.-P.; Yan, Z.-G. A Review of the Toxic Effects of Ammonia on Invertebrates in Aquatic Environments. Environ. Pollut. 2023, 336, 122374. [Google Scholar] [CrossRef] [PubMed]
- USEPA. Update of Ambient Water QualityCriteria for Ammonia; United States Environmental Protection Agency: Washington, DC, USA, 2013; p. 2013. [Google Scholar]
- Vera, L.; Aguilar Galarza, B.; Reinoso, S.; Bohórquez-Cruz, M.; Sonnenholzner, S.; Argüello-Guevara, W. Determination of Acute Toxicity of Unionized Ammonia in Juvenile Longfin Yellowtail (Seriola rivoliana). J. World Aquac. Soc. 2023, 54, 1110–1120. [Google Scholar] [CrossRef]
- van der Meeren, T.; Mangor-Jensen, A. Tolerance of Atlantic Cod (Gadus morhua L.) Larvae to Acute Ammonia Exposure. Aquac. Int. 2020, 28, 1753–1769. [Google Scholar] [CrossRef]
- Almeida, D.B.; Magalhães, C.; Sousa, Z.; Borges, M.T.; Silva, E.; Blanquet, I.; Mucha, A.P. Microbial Community Dynamics in a Hatchery Recirculating Aquaculture System (RAS) of Sole (Solea senegalensis). Aquaculture 2021, 539, 736592. [Google Scholar] [CrossRef]
- Dahle, S.W.; Gaarden, S.I.; Buhaug, J.F.; Netzer, R.; Attramadal, K.J.K.; Busche, T.; Aas, M.; Ribicic, D.; Bakke, I. Long-Term Microbial Community Structures and Dynamics in a Commercial RAS during Seven Production Batches of Atlantic Salmon Fry (Salmo salar). Aquaculture 2023, 565, 739155. [Google Scholar] [CrossRef]
- Neissi, A.; Rafiee, G.; Rahimi, S.; Farahmand, H.; Pandit, S.; Mijakovic, I. Enriched Microbial Communities for Ammonium and Nitrite Removal from Recirculating Aquaculture Systems. Chemosphere 2022, 295, 133811. [Google Scholar] [CrossRef] [PubMed]
- Al-Ajeel, S.; Spasov, E.; Sauder, L.A.; McKnight, M.M.; Neufeld, J.D. Ammonia-Oxidizing Archaea and Complete Ammonia-Oxidizing Nitrospira in Water Treatment Systems. Water Res. X 2022, 15, 100131. [Google Scholar] [CrossRef] [PubMed]
- Ghimire-Kafle, S.; Weaver, M.E.; Kimbrel, M.P.; Bollmann, A. Competition between Ammonia-Oxidizing Archaea and Complete Ammonia Oxidizers from Freshwater Environments. Appl. Environ. Microbiol. 2024, 90, e01698-23. [Google Scholar] [CrossRef] [PubMed]
- Kits, K.D.; Sedlacek, C.J.; Lebedeva, E.V.; Han, P.; Bulaev, A.; Pjevac, P.; Daebeler, A.; Romano, S.; Albertsen, M.; Stein, L.Y.; et al. Kinetic Analysis of a Complete Nitrifier Reveals an Oligotrophic Lifestyle. Nature 2017, 549, 269–272. [Google Scholar] [CrossRef]
- Yin, Q.; Sun, Y.; Li, B.; Feng, Z.; Wu, G. The r/K Selection Theory and Its Application in Biological Wastewater Treatment Processes. Sci. Total Environ. 2022, 824, 153836. [Google Scholar] [CrossRef]
- Kikuchi, S.; Fujitani, H.; Ishii, K.; Isshiki, R.; Sekiguchi, Y.; Tsuneda, S. Characterisation of Bacteria Representing a Novel Nitrosomonas Clade: Physiology, Genomics and Distribution of Missing Ammonia Oxidizer. Environ. Microbiol. Rep. 2023, 15, 404–416. [Google Scholar] [CrossRef]
- Li, H.; Cui, Z.; Cui, H.; Bai, Y.; Yin, Z.; Qu, K. A Review of Influencing Factors on a Recirculating Aquaculture System: Environmental Conditions, Feeding Strategies, and Disinfection Methods. J. World Aquac. Soc. 2023, 54, 566–602. [Google Scholar] [CrossRef]
- Chen, Z.; Chang, Z.; Zhang, L.; Jiang, Y.; Ge, H.; Song, X.; Chen, S.; Zhao, F.; Li, J. Effects of Water Recirculation Rate on the Microbial Community and Water Quality in Relation to the Growth and Survival of White Shrimp (Litopenaeus vannamei). BMC Microbiol. 2019, 19, 192. [Google Scholar] [CrossRef] [PubMed]
- Rieder, J.; Kapopoulou, A.; Bank, C.; Adrian-Kalchhauser, I. Metagenomics and Metabarcoding Experimental Choices and Their Impact on Microbial Community Characterization in Freshwater Recirculating Aquaculture Systems. Environ. Microbiome 2023, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network Analysis Methods for Studying Microbial Communities: A Mini Review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef]
- EHEIM GmbH & Co. KG. EHEIM Professional 3 1200XLT Manual. Available online: https://eheim.com/en_GB/aquatics/technology/external-filters/professionel-3/professionel-3-1200xlt?c=1939 (accessed on 1 October 2024).
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc (accessed on 28 November 2023).
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an Efficient Tool for Accurately Reconstructing Single Genomes from Complex Microbial Communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning Metagenomic Contigs by Coverage and Composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of Genomes from Metagenomes via a Dereplication, Aggregation and Scoring Strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory Friendly Classification with the Genome Taxonomy Database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pan, J.; Zhou, Z.; Wu, J.; Liu, Y.; Lin, J.-G.; Hong, Y.; Li, X.; Li, M.; Gu, J.-D. Complex Microbial Nitrogen-Cycling Networks in Three Distinct Anammox-Inoculated Wastewater Treatment Systems. Water Res. 2020, 168, 115142. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for Distilling Microbial Metabolism to Automate the Curation of Microbiome Function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef] [PubMed]
- Hammer, D.A.T.; Ryan, P.D.; Hammer, Ø.; Harper, D.A.T. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 1. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Burrell, P.C.; Phalen, C.M.; Hovanec, T.A. Identification of Bacteria Responsible for Ammonia Oxidation in Freshwater Aquaria. Appl. Environ. Microbiol. 2001, 67, 5791–5800. [Google Scholar] [CrossRef]
- Bagchi, S.; Vlaeminck, S.E.; Sauder, L.A.; Mosquera, M.; Neufeld, J.D.; Boon, N. Temporal and Spatial Stability of Ammonia-Oxidizing Archaea and Bacteria in Aquarium Biofilters. PLoS ONE 2014, 9, e113515. [Google Scholar] [CrossRef] [PubMed]
- Bartelme, R.P.; McLellan, S.L.; Newton, R.J. Freshwater Recirculating Aquaculture System Operations Drive Biofilter Bacterial Community Shifts around a Stable Nitrifying Consortium of Ammonia-Oxidizing Archaea and Comammox Nitrospira. Front. Microbiol. 2017, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- McKnight, M.M.; Neufeld, J.D. Comammox Nitrospira among Dominant Ammonia Oxidizers within Aquarium Biofilter Microbial Communities. Appl. Environ. Microbiol. 2024, 90, e00104-24. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, Y.; Shu, L.; Gu, H.; Liu, F.; Ding, J.; Zeng, J.; Wang, C.; He, Z.; Xu, M.; et al. Unraveling the Important Role of Comammox Nitrospira to Nitrification in the Coastal Aquaculture System. Front. Microbiol. 2024, 15, 1355859. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.-Y.; Sedlacek, C.J.; Kits, K.D.; Mueller, A.J.; Rhee, S.-K.; Hink, L.; Nicol, G.W.; Bayer, B.; Lehtovirta-Morley, L.; Wright, C.; et al. Ammonia-Oxidizing Archaea Possess a Wide Range of Cellular Ammonia Affinities. ISME J. 2022, 16, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Sakoula, D.; Koch, H.; Frank, J.; Jetten, M.S.M.; van Kessel, M.A.H.J.; Lücker, S. Enrichment and Physiological Characterization of a Novel Comammox Nitrospira Indicates Ammonium Inhibition of Complete Nitrification. ISME J. 2021, 15, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, C.J.; Nielsen, S.; Greis, K.D.; Haffey, W.D.; Revsbech, N.P.; Ticak, T.; Laanbroek, H.J.; Bollmann, A. Effects of Bacterial Community Members on the Proteome of the Ammonia-Oxidizing Bacterium Nitrosomonas Sp. Strain Is79. Appl. Environ. Microbiol. 2016, 82, 4776–4788. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, C.J.; McGowan, B.; Suwa, Y.; Sayavedra-Soto, L.; Laanbroek, H.J.; Stein, L.Y.; Norton, J.M.; Klotz, M.G.; Bollmann, A. A Physiological and Genomic Comparison of Nitrosomonas Cluster 6a and 7 Ammonia-Oxidizing Bacteria. Microb. Ecol. 2019, 78, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Thandar, S.M.; Ushiki, N.; Fujitani, H.; Sekiguchi, Y.; Tsuneda, S. Ecophysiology and Comparative Genomics of Nitrosomonas mobilis Ms1 Isolated from Autotrophic Nitrifying Granules of Wastewater Treatment Bioreactor. Front. Microbiol. 2016, 7, 1869. [Google Scholar] [CrossRef] [PubMed]
- Martens-Habbena, W.; Berube, P.M.; Urakawa, H.; de la Torre, J.R.; Stahl, D.A. Ammonia Oxidation Kinetics Determine Niche Separation of Nitrifying Archaea and Bacteria. Nature 2009, 461, 976–979. [Google Scholar] [CrossRef]
- Roalkvam, I.; Drønen, K.; Dahle, H.; Wergeland, H.I. A Case Study of Biofilter Activation and Microbial Nitrification in a Marine Recirculation Aquaculture System for Rearing Atlantic Salmon (Salmo salar L.). Aquac. Res. 2021, 52, 94–104. [Google Scholar] [CrossRef]
- Bai, X.; Hu, X.; Liu, J.; Yu, Z.; Jin, J.; Liu, X.; Wang, G. Canonical Ammonia Oxidizers and Comammox Clade A Play Active Roles in Nitrification in a Black Soil at Different PH and Ammonium Concentrations. Biol. Fertil. Soils 2024, 60, 471–481. [Google Scholar] [CrossRef]
- Ding, F.; He, T.; Qi, X.; Zhang, H.; An, L.; Xu, S.; Zhang, X. Comammox Nitrospira Dominates the Nitrification in Artificial Coniferous Forest Soils of the Qilian Mountains. Sci. Total Environ. 2024, 906, 167653. [Google Scholar] [CrossRef]
- Hsu, P.-C.; Di, H.J.; Cameron, K.; Podolyan, A.; Chau, H.; Luo, J.; Miller, B.; Carrick, S.; Johnstone, P.; Ferguson, S.; et al. Comammox Nitrospira Clade B Is the Most Abundant Complete Ammonia Oxidizer in a Dairy Pasture Soil and Inhibited by Dicyandiamide and High Ammonium Concentrations. Front. Microbiol. 2022, 13, 1048735. [Google Scholar] [CrossRef]
- Yuan, D.; Zheng, L.; Tan, Q.; Wang, X.; Xing, Y.; Wang, H.; Wang, S.; Zhu, G. Comammox Activity Dominates Nitrification Process in the Sediments of Plateau Wetland. Water Res. 2021, 206, 117774. [Google Scholar] [CrossRef] [PubMed]
- Steuernagel, L.; de Léon Gallegos, E.L.; Azizan, A.; Dampmann, A.-K.; Azari, M.; Denecke, M. Availability of Carbon Sources on the Ratio of Nitrifying Microbial Biomass in an Industrial Activated Sludge. Int. Biodeterior. Biodegrad. 2018, 129, 133–140. [Google Scholar] [CrossRef]
- Navada, S.; Knutsen, M.F.; Bakke, I.; Vadstein, O. Nitrifying Biofilms Deprived of Organic Carbon Show Higher Functional Resilience to Increases in Carbon Supply. Sci. Rep. 2020, 10, 7121. [Google Scholar] [CrossRef] [PubMed]
- Hüpeden, J.; Wemheuer, B.; Indenbirken, D.; Schulz, C.; Spieck, E. Taxonomic and Functional Profiling of Nitrifying Biofilms in Freshwater, Brackish and Marine RAS Biofilters. Aquac. Eng. 2020, 90, 102094. [Google Scholar] [CrossRef]
- Sunish, K.S.; Sreedharan, K.; Shadha Nazreen, S.K. Actinomycetes as a Promising Candidate Bacterial Group for the Health Management of Aquaculture Systems: A Review. Rev. Aquac. 2023, 15, 1198–1226. [Google Scholar] [CrossRef]
- Babu, D.T.; Archana, K.; Kachiprath, B.; Solomon, S.; Jayanath, G.; Singh, I.S.B.; Philip, R. Marine Actinomycetes as Bioremediators in Penaeus Monodon Rearing System. Fish Shellfish Immunol. 2018, 75, 231–242. [Google Scholar] [CrossRef]
- Albuquerque, L.; França, L.; Rainey, F.A.; Schumann, P.; Nobre, M.F.; da Costa, M.S. Gaiella occulta Gen. Nov., Sp. Nov., a Novel Representative of a Deep Branching Phylogenetic Lineage within the Class Actinobacteria and Proposal of Gaiellaceae Fam. Nov. and Gaiellales Ord. Nov. Syst. Appl. Microbiol. 2011, 34, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, M.; Francisco, I.; Díaz-González, F.; Diaz, M.; Quatrini, R.; Beamud, G.; Pedrozo, F.; Temporetti, P. Nutrient Structure Dynamics and Microbial Communities at the Water–Sediment Interface in an Extremely Acidic Lake in Northern Patagonia. Front. Microbiol. 2024, 15, 1335978. [Google Scholar] [CrossRef]
- Liu, M.; Huang, H.; Bao, S.; Tong, Y. Microbial Community Structure of Soils in Bamenwan Mangrove Wetland. Sci. Rep. 2019, 9, 8406. [Google Scholar] [CrossRef]
- Chen, R.-W.; He, Y.-Q.; Cui, L.-Q.; Li, C.; Shi, S.-B.; Long, L.-J.; Tian, X.-P. Diversity and Distribution of Uncultured and Cultured Gaiellales and Rubrobacterales in South China Sea Sediments. Front. Microbiol. 2021, 12, 657072. [Google Scholar] [CrossRef] [PubMed]
- Severino, R.; Froufe, H.J.C.; Barroso, C.; Albuquerque, L.; Lobo-da-Cunha, A.; da Costa, M.S.; Egas, C. High-Quality Draft Genome Sequence of Gaiella occulta Isolated from a 150 Meter Deep Mineral Water Borehole and Comparison with the Genome Sequences of Other Deep-Branching Lineages of the Phylum Actinobacteria. Microbiologyopen 2019, 8, e00840. [Google Scholar] [CrossRef] [PubMed]
- Burut-Archanai, S.; Ubertino, D.; Chumtong, P.; Mhuantong, W.; Powtongsook, S.; Piyapattanakorn, S. Dynamics of Microbial Community During Nitrification Biofilter Acclimation with Low and High Ammonia. Mar. Biotechnol. 2021, 23, 671–681. [Google Scholar] [CrossRef]
- Deng, M.; Dai, Z.; Senbati, Y.; Li, L.; Song, K.; He, X. Aerobic Denitrification Microbial Community and Function in Zero-Discharge Recirculating Aquaculture System Using a Single Biofloc-Based Suspended Growth Reactor: Influence of the Carbon-to-Nitrogen Ratio. Front. Microbiol. 2020, 11, 1760. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hasezawa, R.; Saito, R.; Okano, K.; Shimizu, K.; Utsumi, M. Abundance and Diversity of Nitrifying Microorganisms in Marine Recirculating Aquaculture Systems. Water 2022, 14, 2744. [Google Scholar] [CrossRef]
- Bayer, B.; Pelikan, C.; Bittner, M.J.; Reinthaler, T.; Könneke, M.; Herndl, G.J.; Offre, P. Proteomic Response of Three Marine Ammonia-Oxidizing Archaea to Hydrogen Peroxide and Their Metabolic Interactions with a Heterotrophic Alphaproteobacterium. mSystems 2019, 4, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Cremin, K.; Duxbury, S.J.N.; Rosko, J.; Soyer, O.S. Formation and Emergent Dynamics of Spatially Organized Microbial Systems. Interface Focus 2023, 13, 20220062. [Google Scholar] [CrossRef]
- Xu, S.; Chai, W.; Xiao, R.; Smets, B.F.; Palomo, A.; Lu, H. Survival Strategy of Comammox Bacteria in a Wastewater Nutrient Removal System with Sludge Fermentation Liquid as Additional Carbon Source. Sci. Total Environ. 2022, 802, 149862. [Google Scholar] [CrossRef] [PubMed]
- Mehrani, M.-J.; Sobotka, D.; Kowal, P.; Guo, J.; Mąkinia, J. New Insights into Modeling Two-Step Nitrification in Activated Sludge Systems—The Effects of Initial Biomass Concentrations, Comammox and Heterotrophic Activities. Sci. Total Environ. 2022, 848, 157628. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before Feeding I (BF_I) | After Feeding I (AF_I) | Before Feeding II (BF_II) | After Feeding II (AF_II) | ||
|---|---|---|---|---|---|
| Ammonia (mg N/L) | inflow | 0.01 | 0.016 | 0.007 | 0.008 |
| outflow | 0.011 | 0.011 | 0.007 | 0.009 | |
| Nitrite (mg N/L) | inflow | 0.015 | 0.009 | 0.065 | 0.01 |
| outflow | 0.014 | 0.011 | 0.06 | 0.011 | |
| Nitrate (mg N/L) | inflow | 4.05 | 4.66 | 4.39 | 2.49 |
| outflow | 4.33 | 4.56 | 4.37 | 4.41 | |
| Dissolved oxygen (mg/L) | 8.96 | 9.02 | 8.95 | 9.07 | |
| Temperature (°C) | 19.1 | 19.2 | 19.7 | 19.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godzieba, M.; Hliwa, P.; Ciesielski, S. Network of Nitrifying Bacteria in Aquarium Biofilters: An Unfaltering Cooperation Between Comammox Nitrospira and Ammonia-Oxidizing Archaea. Water 2025, 17, 52. https://doi.org/10.3390/w17010052
Godzieba M, Hliwa P, Ciesielski S. Network of Nitrifying Bacteria in Aquarium Biofilters: An Unfaltering Cooperation Between Comammox Nitrospira and Ammonia-Oxidizing Archaea. Water. 2025; 17(1):52. https://doi.org/10.3390/w17010052
Chicago/Turabian StyleGodzieba, Martyna, Piotr Hliwa, and Slawomir Ciesielski. 2025. "Network of Nitrifying Bacteria in Aquarium Biofilters: An Unfaltering Cooperation Between Comammox Nitrospira and Ammonia-Oxidizing Archaea" Water 17, no. 1: 52. https://doi.org/10.3390/w17010052
APA StyleGodzieba, M., Hliwa, P., & Ciesielski, S. (2025). Network of Nitrifying Bacteria in Aquarium Biofilters: An Unfaltering Cooperation Between Comammox Nitrospira and Ammonia-Oxidizing Archaea. Water, 17(1), 52. https://doi.org/10.3390/w17010052