
Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Model in Immunotherapy Research

 ,
,
Abstract
:1. Introduction
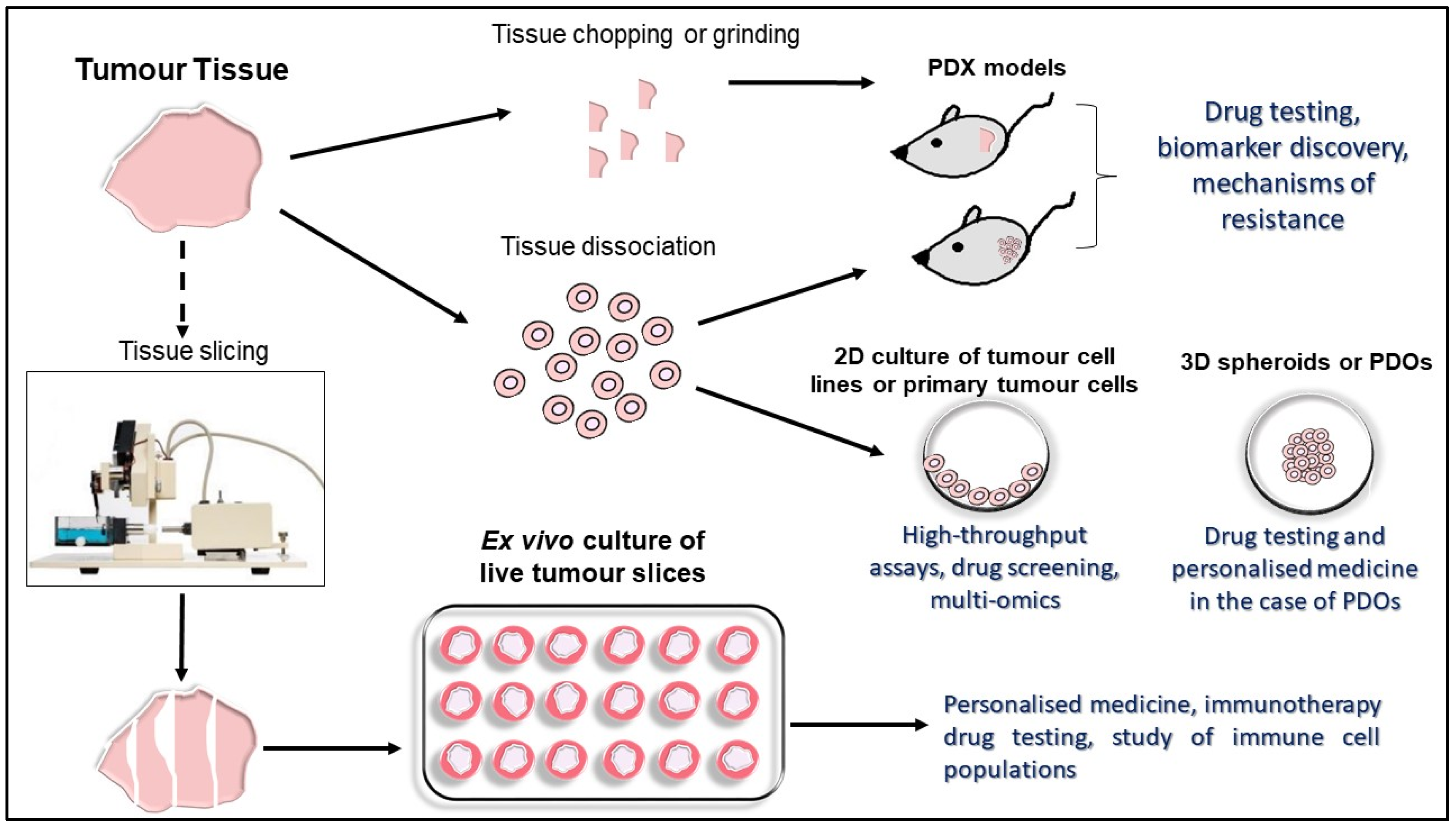
2. Established In Vitro, In Vivo and Ex Vivo Experimental Tumor Models
2.1. Three-Dimensional Tumor Cell Cultures
2.2. Three-Dimensional Human Tumor Models
2.3. PDX Murine Models
3. Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Platform in Immunotherapy Research
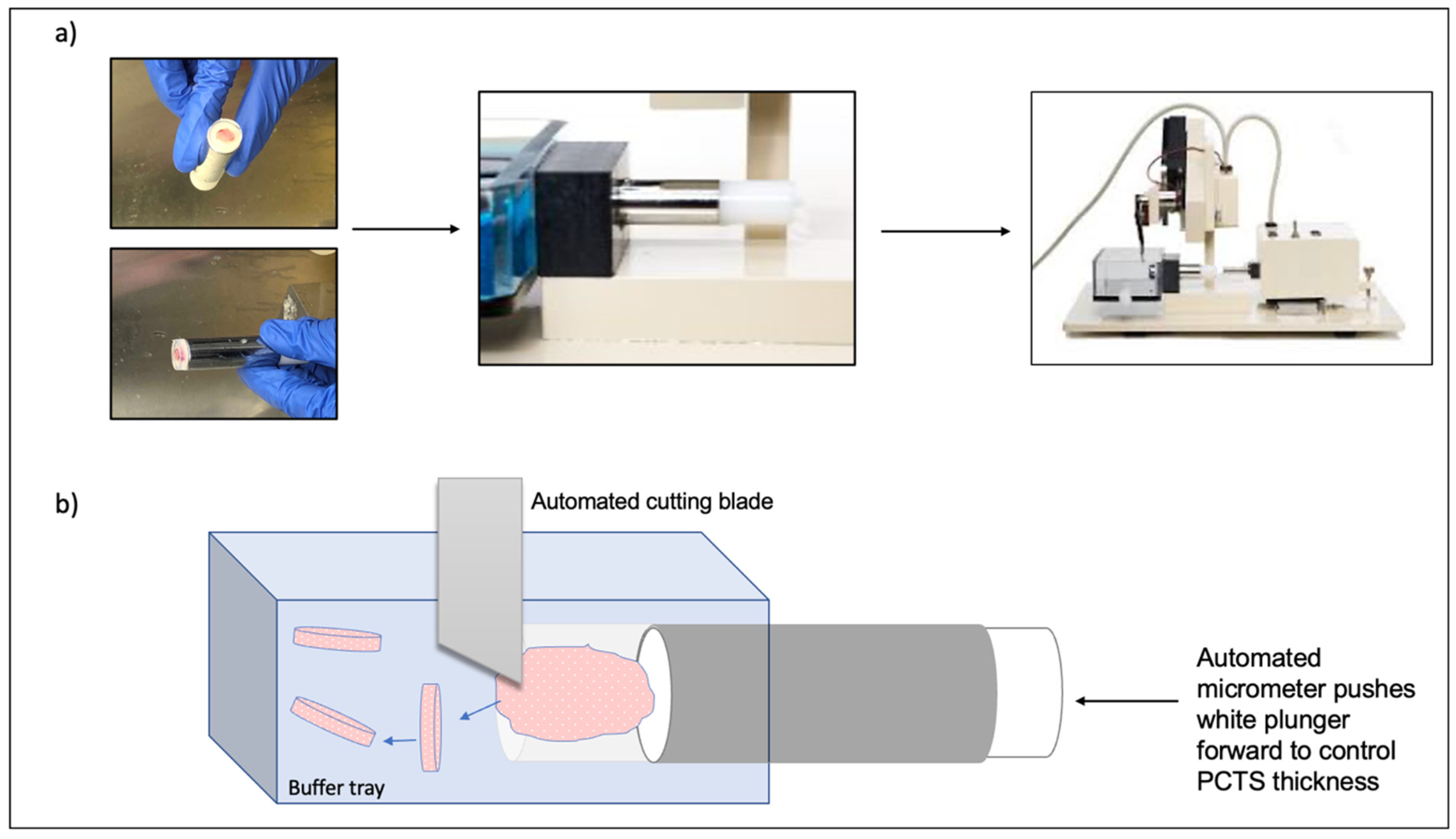
3.1. Vibratome Technology for PCTS Generation
3.2. Generation and Culture of PCTS
4. What Is the Value of PCTS in Immunotherapy Research?
4.1. PCTS as a Model for Studying Localisation and Function of the Immune TME
4.2. PCTS as a Platform to Assess Chimeric Antigen Receptor (CAR) T Cell Infiltration and Activation
4.3. Use of PCTS in Predicting Patient Response to Immunotherapy
5. PCTS: Advantages and Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2–9. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Futur. Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumar, R.; Chan, M.; Shin, J.S.; Nishida-Aoki, N.; Kenerson, H.L.; Elemento, O.; Beltran, H.; Yeung, R.; Gujral, T.S. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. OncoImmunology 2019, 8, e1670019. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Gujral, T.S. Emerging approaches to study cell-cell interactions in tumor microenvironment. Oncotarget 2019, 10, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Chow, A.B.; Mattingly, R.R. Three-Dimensional Overlay Culture Models of Human Breast Cancer Reveal a Critical Sensitivity to Mitogen-Activated Protein Kinase Kinase Inhibitors. J. Pharmacol. Exp. Ther. 2009, 332, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Law, A.M.K.; de la Fuente, L.R.; Grundy, T.J.; Fang, G.; Valdes-Mora, F.; Gallego-Ortega, D. Advancements in 3D Cell Culture Systems for Personalizing Anti-Cancer Therapies. Front. Oncol. 2021, 11, 782766. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, A.A.; Li, E.; Weiner, L.M. 3D Culture Systems for Exploring Cancer Immunology. Cancers 2020, 13, 56. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, X.; Ding, R.; Yang, L.; Lyu, X.; Zeng, J.; Lei, J.H.; Wang, L.; Bi, J.; Shao, N.; et al. Patient-Derived Organoids Can Guide Personalized-Therapies for Patients with Advanced Breast Cancer. Adv. Sci. 2021, 8, 2101176. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.-J.; Chun, S.-M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef]
- Seidlitz, T.; Koo, B.-K.; Stange, D.E. Gastric organoids—An in vitro model system for the study of gastric development and road to personalized medicine. Cell Death Differ. 2020, 28, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.; Kryeziu, K.; Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Langerud, J.; Røsok, B.I.; Totland, M.Z.; Brunsell, T.H.; Pellinen, T.; et al. Patient-Derived Organoids from Multiple Colorectal Cancer Liver Metastases Reveal Moderate Intra-patient Pharmacotranscriptomic Heterogeneity. Clin. Cancer Res. 2020, 26, 4107–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Sun, L.; Liu, M.; Mao, Y. Patient-derived organoids: A promising model for personalized cancer treatment. Gastroenterol. Rep. 2018, 6, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models—The future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Sullivan, K.M.; Seo, Y.D.; Stadeli, K.M.; Ussakli, C.; Yan, X.; Lausted, C.; Pillarisetty, V.G.; Park, J.O.; Riehle, K.J.; et al. Tumor slice culture as a biologic surrogate of human cancer. Ann. Transl. Med. 2020, 8, 114. [Google Scholar] [CrossRef]
- Jiang, X.; Seo, Y.D.; Chang, H.J.; Coveler, A.; Nigjeh, E.N.; Pan, S.; Jalikis, F.; Yeung, R.S.; Crispe, I.N.; Pillarisetty, V.G.; et al. Long-lived pancreatic ductal adenocarcinoma slice cultures enable precise study of the immune microenvironment. Oncoimmunology 2017, 6, e1333210. [Google Scholar] [CrossRef] [Green Version]
- Kenerson, H.L.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G.; Yeung, R.S. Protocol for tissue slice cultures from human solid tumors to study therapeutic response. STAR Protoc. 2021, 2, 100574. [Google Scholar] [CrossRef]
- Mohammed, F.; Arishiya, T.F.; Mohamed, A. Microtomes and microtome knives—A Review and Proposed Classification. Ann. Dent. 2012, 19, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Krumdieck, C.L. Development of a live tissue microtome: Reflections of an amateur machinist. Xenobiotica 2013, 43, 2–7. [Google Scholar] [CrossRef]
- Merz, F.; Gaunitz, F.; Dehghani, F.; Renner, C.; Meixensberger, J.; Gutenberg, A.; Giese, A.; Schopow, K.; Hellwig, C.; Schäfer, M.; et al. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neuro-Oncol. 2013, 15, 670–681. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lampe, J.; Lange, S.; Smirnow, I.; Königsrainer, A.; Hann-Von-Weyhern, C.; Fend, F.; Gregor, M.; Bitzer, M.; Lauer, U.M. Improved reproducibility in preparing precision-cut liver tissue slices. Cytotechnology 2009, 61, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Abdelaal, H.M.; Kim, H.O.; Wagstaff, R.; Sawahata, R.; Southern, P.J.; Skinner, P.J. Comparison of Vibratome and Compresstome sectioning of fresh primate lymphoid and genital tissues for in situ MHC-tetramer and immunofluorescence staining. Biol. Proced. Online 2015, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Inc, P.I. Compresstome Blog Article #3: Benefits of Compression. 2020. Available online: https://www.precisionary.com/our-slicers/compresstome-blog-article-3-benefits-of-compression/ (accessed on 23 January 2022).
- Iulianella, A. Cutting Thick Sections Using a Vibratome. Cold Spring Harb. Protoc 2017. [Google Scholar] [CrossRef] [PubMed]
- Roelants, C.; Pillet, C.; Franquet, Q.; Sarrazin, C.; Peillerson, N.; Giacosa, S.; Guyon, L.; Fontanell, A.; Fiard, G.; Long, J.-A.; et al. Ex-Vivo Treatment of Tumor Tissue Slices as a Predictive Preclinical Method to Evaluate Targeted Therapies for Patients with Renal Carcinoma. Cancers 2020, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Bougherara, H.; Barrin, S.; Mansuet-Lupo, A.; Alifano, M.; Damotte, D.; Donnadieu, E. Ex Vivo Imaging of Resident CD8 T Lymphocytes in Human Lung Tumor Slices Using Confocal Microscopy. J. Vis. Exp. 2017, 130, 55709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, R.; Lapshyna, O.; Eckelmann, S.; Honselmann, K.; Bolm, L.; Winkel, M.T.; Deichmann, S.; Schilling, O.; Kruse, C.; Keck, T.; et al. Organotypic Slice Cultures as Preclinical Models of Tumor Microenvironment in Primary Pancreatic Cancer and Metastasis. J. Vis. Exp. 2021, 172. [Google Scholar] [CrossRef] [PubMed]
- de Hoogt, R.; Estrada, M.F.; Vidic, S.; Davies, E.J.; Osswald, A.; Barbier, M.; Santo, V.E.; Gjerde, K.; van Zoggel, H.J.A.A.; Blom, S.; et al. Protocols and characterization data for 2D, 3D, and slice-based tumor models from the PREDECT project. Sci. Data 2017, 4, 170170. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.J.; Lizarraga, M.; Waziri, A.; Foshay, K.M. A Human Glioblastoma Organotypic Slice Culture Model for Study of Tumor Cell Migration and Patient-specific Effects of Anti-Invasive Drugs. J. Vis. Exp. 2017, 125, 53557. [Google Scholar] [CrossRef]
- Majorova, D.; Atkins, E.; Martineau, H.; Vokral, I.; Oosterhuis, D.; Olinga, P.; Wren, B.; Cuccui, J.; Werling, D. Use of Precision-Cut Tissue Slices as a Translational Model to Study Host-Pathogen Interaction. Front. Veter Sci. 2021, 8, 686088. [Google Scholar] [CrossRef]
- Alsafadi, H.N.; Uhl, F.E.; Pineda, R.H.; Bailey, K.E.; Rojas, M.; Wagner, D.E.; Königshoff, M. Applications and Approaches for Three-Dimensional Precision-Cut Lung Slices. Disease Modeling and Drug Discovery. Am. J. Respir. Cell Mol. Biol. 2020, 62, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.R.; Tiriac, H.; Mendoza, T.H.; Wascher, A.; Lowy, A.M. Using Organotypic Tissue Slices to Investigate the Microenvironment of Pancreatic Cancer: Pharmacotyping and Beyond. Cancers(Basel) 2021, 13, 4991. [Google Scholar] [CrossRef]
- Kishan, A.T.N.; Nicole, S.V.; Humberto, S.; van Deurzen Carolien, H.M.; den Bakker Michael, A.; Jan, H.J.H.; Roland, K.; Vreeswijk Maaike, P.G.; Agnes, J.; van Gent, D.C. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 2016, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.J.; Dong, M.; Gutekunst, M.; Närhi, K.; Van Zoggel, H.J.A.A.; Blom, S.; Nagaraj, A.; Metsalu, T.; Oswald, E.; Erkens-Schulze, S.; et al. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci. Rep. 2015, 5, 17187. [Google Scholar] [CrossRef]
- Boutet, M.; Gauthier, L.; Leclerc, M.; Gros, G.; de Montpreville, V.; Théret, N.; Donnadieu, E.; Mami-Chouaib, F. TGFbeta Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 1757–1769. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantari-Mimoun, C.; Barrin, S.; Haghiri, S.; Haghri, S.; Gervais, C.; Mittelsttaet, J.; Mockel-Tenbrinck, N.; Kinkhabwala, A.; Damotte, D.; Lupo, A.; et al. CAR T-cell Entry into Tumor Islets Is a Two-Step Process Dependent on IFNgamma and ICAM-1. Cancer Immunol. Res. 2021, 9, 1425–1438. [Google Scholar] [CrossRef]
- Melssen, M.M.; Lindsay, R.S.; Stasiak, K.; Rodriguezi, A.B.; Briegel, A.M.; Cyranowski, S.; Rutkowski, M.R.; Conaway, M.R.; Melief, C.J.M.; van der Burg, S.H.; et al. Differential Expression of CD49a and CD49b Determines Localization and Function of Tu-mor-Infiltrating CD8(+) T Cells. Cancer Immunol. Res. 2021, 9, 583–597. [Google Scholar] [CrossRef]
- Park, S.; Gebhardt, T.; Mackay, L.K. Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol. 2019, 40, 735–747. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; O’Brien, S.M.; Thompson, J.C.; Rao, A.S.; Stadanlick, J.E.; Martinez, M.; Liousia, M.; Cantu, E.; Cengel, K.; Moon, E.K.; et al. Phenotypic and functional analysis of malignant mesothelioma tumor-infiltrating lymphocytes. OncoImmunology 2019, 8, e1638211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Shen, Y.; Luyten, S.; Yang, Y.; Jiang, X. Tissue-resident memory CD8+ T cells in cancer immunology and immunotherapy. Pharmacol. Res. 2020, 159, 104876. [Google Scholar] [CrossRef]
- Byrne, A.; Savas, P.; Sant, S.; Li, R.; Virassamy, B.; Luen, S.J.; Beavis, P.; Mackay, L.K.; Neeson, P.J.; Loi, S. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nat. Rev. Clin. Oncol. 2020, 17, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngô, C.; Damotte, D.; Le Frère-Belda, M.-A.; Donnadieu, E.; Peranzoni, E. Real-Time Imaging of Resident T Cells in Human Lung and Ovarian Carcinomas Reveals How Different Tumor Microenvironments Control T Lymphocyte Migration. Front. Immunol. 2015, 6, 500. [Google Scholar] [CrossRef] [Green Version]
- Roife, D.; Dai, B.; Kang, Y.; Perez, M.V.R.; Pratt, M.; Li, X.; Fleming, J.B. Ex Vivo Testing of Patient-Derived Xenografts Mirrors the Clinical Outcome of Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2016, 22, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Philippi, C.; Loretz, B.; Nafee, N.; Schaefer, U.F.; Friedel, G.; Ammon-Treiber, S.; Griese, E.-U.; Lehr, C.-M.; Klotz, U.; et al. Tissue slice model of human lung cancer to investigate telomerase inhibition by nanoparticle delivery of antisense 2’-O-methyl-RNA. Int. J. Pharm. 2011, 419, 33–42. [Google Scholar] [CrossRef]
- Zhang, W.; van Weerden, W.M.; de Ridder, C.M.A.; Erkens-Schulze, S.; Schönfeld, E.; Meijer, T.G.; Kanaar, R.; van Gent, D.C.; Nonnekens, J. Ex vivo treatment of prostate tumor tissue recapitulates in vivo therapy response. Prostate 2019, 79, 390–402. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.Z.; Wagner, D.C.; Hörner, N.; Horst, D.; Lang, H.; Tagscherer, K.E.; Roth, W. Ex vivo tissue slice culture system to measure drug-response rates of hepatic metastatic colorectal cancer. BMC Cancer 2019, 19, 1030. [Google Scholar] [CrossRef]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignail, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8(+) T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, L.F.; Rodriguez, A.D.; Dereli-Korkut, Z.; Lin, R.; Castro, K.; Mikheev, A.; Monnat, R.J.; Folch, A.; Rostomily, R.C. Multiplexed drug testing of tumor slices using a microfluidic platform. NPJ Precis Oncol 2020, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Stegmayr, J.; Wagner, D.E. The dawn of the omics era in human precision-cut lung slices. Eur. Respir. J. 2021, 58, 2100203. [Google Scholar] [CrossRef]
- Khan, M.M.; Poeckel, D.; Halavatyi, A.; Zukowska-Kasprzyk, J.; Stein, F.; Vappiani, J.; Sevin, D.C.; Tischer, C.; Zinn, N.; Eley, J.D.; et al. An integrated multiomic and quantitative label-free microscopy-based approach to study profibrotic signalling in ex vivo human precision-cut lung slices. Eur. Respir J. 2021, 58, 2000221. [Google Scholar] [CrossRef] [PubMed]
- Stegmayr, J.; Alsafadi, H.N.; Langwiński, W.; Niroomand, A.; Lindstedt, S.; Leigh, N.D.; Wagner, D.E. Isolation of high-yield and -quality RNA from human precision-cut lung slices for RNA-sequencing and computational integration with larger patient cohorts. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L232–L240. [Google Scholar] [CrossRef]
- Fan, T.W.; Higashi, R.M.; Song, H.; Daneshmandi, S.; Mahan, A.L.; Purdom, M.S.; Bocklage, T.J.; Pittman, T.A.; He, D.; Wang, C.; et al. Innate immune activation by checkpoint inhibition in human patient-derived lung cancer tissues. eLife 2021, 10, e69578. [Google Scholar] [CrossRef]
- Bigaeva, E.; Gore, E.; Simon, E.; Zwick, M.; Oldenburger, A.; De Jong, K.P.; Hofker, H.S.; Schlepütz, M.; Nicklin, P.; Boersema, M.; et al. Transcriptomic characterization of culture-associated changes in murine and human precision-cut tissue slices. Arch. Toxicol. 2019, 93, 3549–3583. [Google Scholar] [CrossRef] [Green Version]
- Ghaderi, M.; Moro, C.F.; Elduayen, S.P.; Hultin, E.; Verbeke, C.S.; Björnstedt, M.; Dillner, J. Genome-wide transcriptome profiling of ex-vivo precision-cut slices from human pancreatic ductal adenocarcinoma. Sci. Rep. 2020, 10, 9070. [Google Scholar] [CrossRef]
- Heimbürger, M.; Lärfars, G.; Bratt, J. Prednisolone inhibits cytokine-induced adhesive and cytotoxic interactions between endothelial cells and neutrophils in vitro. Clin. Exp. Immunol. 2000, 119, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Mathema, V.B.; Koh, Y.-S.; Thakuri, B.C.; Sillanpää, M. Parthenolide, a Sesquiterpene Lactone, Expresses Multiple Anti-cancer and Anti-inflammatory Activities. Inflammation 2012, 35, 560–565. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolu-tion of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Microtome Type | Tissues Optimally Sliced with Specific Microtome | Tissue Thickness | Advantages |
|---|---|---|---|
| Saw microtome | Hard specimens, such as teeth and bones | 30 μm or higher | |
| Sledge microtome | Embedded samples | 1–60 μm | |
| Rotary microtome | Thin, embedded samples (manual control) | 0.5–60 µm | |
| Laser microtome | All types of samples | 1 μm or higher | |
| Cryomicrotome | Frozen samples | 2–50 μm | |
| Ultramicrotome | Extremely thin tissue slices | 20–150 nm | Use with specialty microtomes |
| Krumdieck microtome (type of rotary microtome) | PCTS | 100–500 μm | First microtome to be routinely used for PCTS generation. Best for glioblastoma PCTS |
| Vibrating microtome(Vibratome) | Fixed and PCTS | Fixed: >10 μm PCTS: 30–1000 μm |
|
| Compresstome | Fixed and PCTS | Fixed: >10 μm PCTS: 30–1000 μm |
|
| Advantages | Limitations |
|---|---|
| Retain the 3D architecture of the original tumor, including stromal and immune cell compartments. | Lack of vascularization does not permit long-term culture |
| Quick, easy, and relatively inexpensive to generate and culture | Cannot currently be frozen or biobanked |
| Allow studies on the immunobiology of tumors | Any experiments need to fit within the short-term culture timeframe of ~one week |
| Drug screening of immunotherapeutic agents is possible | Low throughput platform with no direct multi-omics possible |
| Permit several assay applications following culture (IHC, flow cytometry, confocal, microscopy, sequencing and supernatant readouts) | Inter- and intra-tumoral heterogeneity observed |
| Organ/tumor-specific transcriptional changes observed following slicing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimou, P.; Trivedi, S.; Liousia, M.; D'Souza, R.R.; Klampatsa, A. Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Model in Immunotherapy Research. Antibodies 2022, 11, 26. https://doi.org/10.3390/antib11020026
Dimou P, Trivedi S, Liousia M, D'Souza RR, Klampatsa A. Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Model in Immunotherapy Research. Antibodies. 2022; 11(2):26. https://doi.org/10.3390/antib11020026
Chicago/Turabian StyleDimou, Paraskevi, Sumita Trivedi, Maria Liousia, Reena R. D'Souza, and Astero Klampatsa. 2022. "Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Model in Immunotherapy Research" Antibodies 11, no. 2: 26. https://doi.org/10.3390/antib11020026