
FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Generalization of the Process
3. Phagocytosis and Trogocytosis Occur in Patients with CLL Treated with Anti-CD20 mAbs
4. Antibody Mediated Immunosuppression/Antigenic Modulation on E
5. NK Cells Can Act as Donors or Acceptors in Trogocytosis
6. Trogocytosis in HIV Disease
7. Daratumumab Mediates Trogocytosis of CD38 on Numerous Cells
8. Antibody Drug Conjugates (ADC)
9. Variations in the Outcome of FcγR-Mediated Transfer Reactions including Results with Epidermal Growth Factor Receptor (EGFR, HER2)
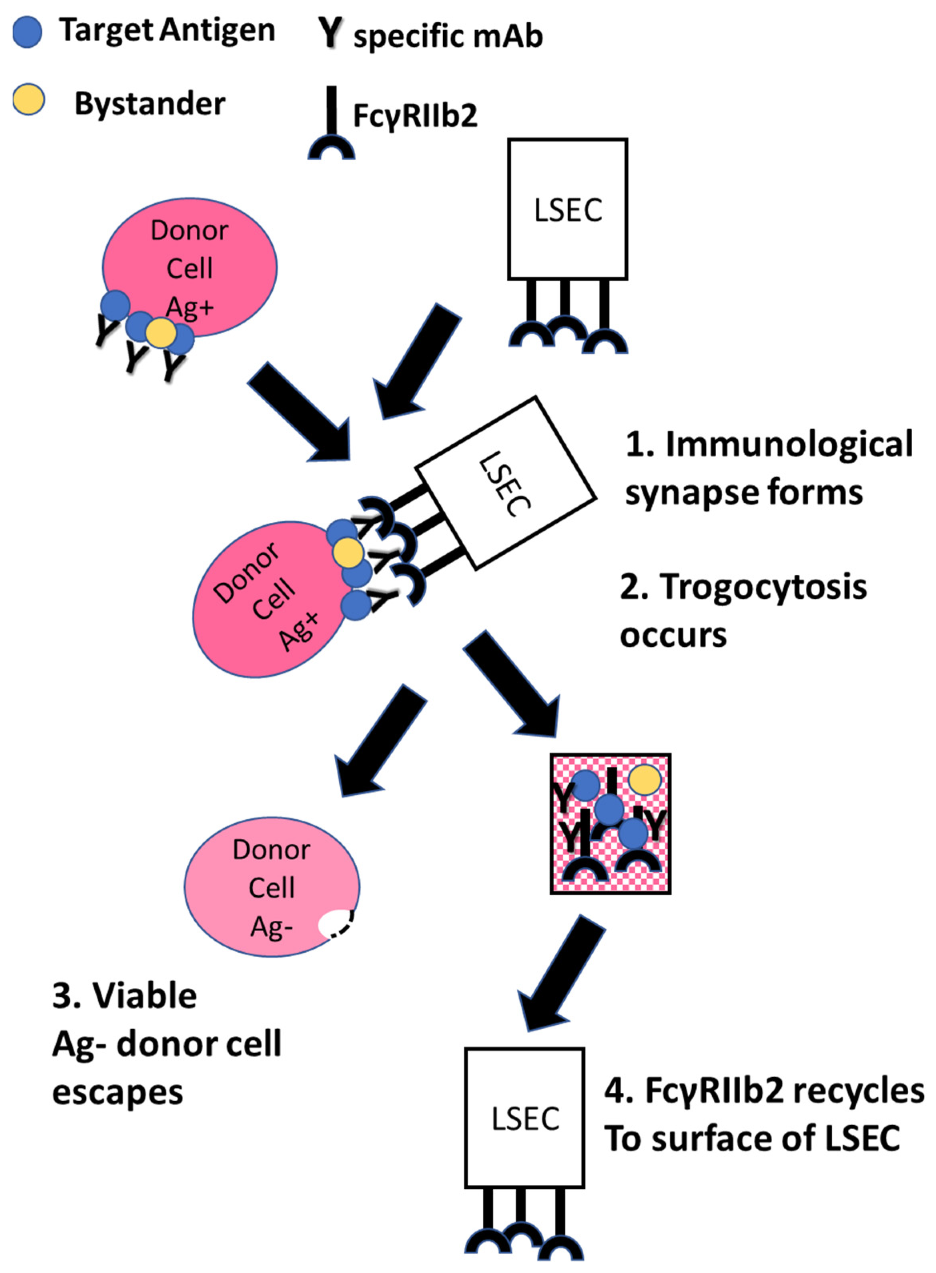
10. The Case for FcγRIIb2
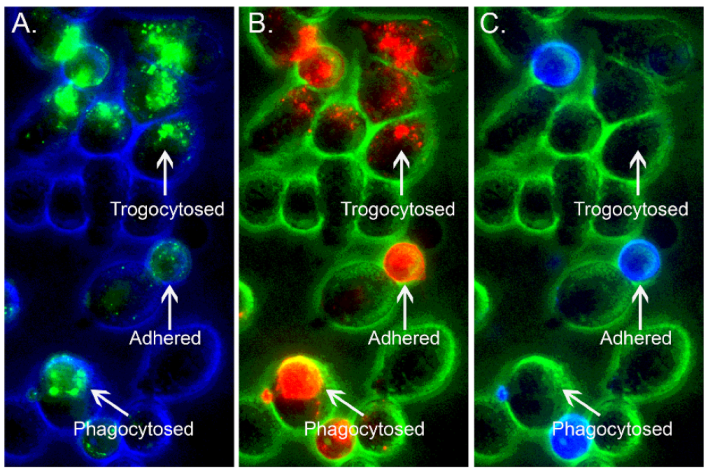
11. Back to the Future: Re-Evaluation of the Immune Adherence Phenomenon/Trogocytosis in a Mouse Model
12. Other Possible Mechanisms
13. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADC | antibody-drug conjugate |
| CLL | chronic lymphocytic leukemia |
| CR1 | complement receptor 1, CD35 |
| DARA | daratumumab, specific for CD38 |
| E | erythrocyte |
| EBV | Epstein–Barr virus |
| FcγR | Fcγ receptor |
| HEL | hen egg lysozyme peptide |
| LSEC | liver sinusoidal endothelial cells |
| MM | multiple myeloma |
| OFA | ofatumumab |
| RTX | rituximab |
| TRA | trastuzumab |
| VP | viral particle |
References
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Müller, R.; von Strandmann, E.P. Antigen Loss Variants: Catching Hold of Escaping Foes. Front. Immunol. 2017, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Schwartz, H. The role of trogocytosis in immune surveillance of Hodgkin lymphoma. Oncoimmunology 2020, 9, 1781334. [Google Scholar] [CrossRef] [PubMed]
- Bettadapur, A.; Miller, H.W.; Ralston, K.S. Biting off What Can Be Chewed: Trogocytosis in Health, Infection, and Disease. Infect. Immun. 2020, 88, e00930-19. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Karasuyama, H. The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells 2021, 10, 1255. [Google Scholar] [CrossRef]
- Nakayama, M.; Hori, A.; Toyoura, S.; Yamaguchi, S.-I. Shaping of T Cell Functions by Trogocytosis. Cells 2021, 10, 1155. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, L.; Xiang, S.; Hu, Y.; Wu, Z.; Shen, J. Gnawing Between Cells and Cells in the Immune System: Friend or Foe? A Review of Trogocytosis. Front. Immunol. 2022, 13, 791006. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. The Multiple Roles of Trogocytosis in Immunity, the Nervous System, and Development. BioMed Res. Int. 2021, 2021, 1601565. [Google Scholar] [CrossRef]
- Dance, A. Core Concept: Cells nibble one another via the under-appreciated process of trogocytosis. Proc. Natl. Acad. Sci. USA 2019, 116, 17608–17610. [Google Scholar] [CrossRef]
- Reed, J.; Reichelt, M.; Wetzel, S. Lymphocytes and Trogocytosis-Mediated Signaling. Cells 2021, 10, 1478. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Greenman, R.; Pizem, Y.; Haus-Cohen, M.; Horev, G.; Denkberg, G.; Shen-Orr, S.; Rubinstein, J.; Reiter, Y. Phenotypic Models of CAR T-Cell Activation Elucidate the Pivotal Regulatory Role of CAR Downmodulation. Mol. Cancer Ther. 2021, 20, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Zhang, Z.; Ren, Z.; Tang, F.; Li, Y. Obstacles and Coping Strategies of CAR-T Cell Immunotherapy in Solid Tumors. Front. Immunol. 2021, 12, 687822. [Google Scholar] [CrossRef]
- Tekguc, M.; Wing, J.B.; Osaki, M.; Long, J.; Sakaguchi, S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releas-ing free PD-L1 on antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2023739118. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.A. The immune-adherence phenomenon. An immunologically specific reaction between microorganisms and erythrocytes leading to enhanced phagocytosis. Science 1953, 118, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.A. The immune-adherence phenomenon. A hypothetical role of erythrocytes in defense against bacteria and virus-es. Proc. Royal. Soc. Med. 1955, 49, 55–58. [Google Scholar] [CrossRef]
- Fearon, D.T. Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J. Exp. Med. 1980, 152, 20–30. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Kohl, J.; Taylor, R.P. Interactions between the complement system and Fcγ receptors. In Antibody Fc: Linking Adaptive and Innate Immunity; Ackerman, M.E., Nimmerjahn, F., Eds.; Elsevier: Philadelphia, PA, USA, 2014; pp. 49–74. [Google Scholar]
- Taylor, R.P.; Lindorfer, M.A. FcγR-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Sporstøl, M.; Fladeby, C.; Kjeken, R.; Barois, N.; Berg, T. Receptor-mediated endocytosis of immune complexes in rat liver sinusoidal endothelial cells is mediated by FcγRIIb2. Hepatology 2007, 46, 871–884. [Google Scholar] [CrossRef]
- Ganesan, L.P.; Kim, J.; Wu, Y.; Mohanty, S.; Phillips, G.S.; Birmingham, D.J.; Robinson, J.M.; Anderson, C.L. FcγRIIb on Liver Sinusoidal Endothelium Clears Small Immune Complexes. J. Immunol. 2012, 189, 4981–4988. [Google Scholar] [CrossRef]
- Abuqayyas, L.; Xhang, X.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcγR on the pharmacokinetics and anti-platelet effects of MWReg30, a monoclonal anti-GBIIb antibody. Int. J. Pharm. 2013, 444, 185–192. [Google Scholar] [CrossRef]
- Anderson, C.L.; Ganesan, L.P.; Robinson, J.M. The biology of the classical Fcγ receptors in non-hematopoietic cells. Immunol. Rev. 2015, 268, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Schölzel, K.; Schildberg, F.A.; Welz, M.; Börner, C.; Geiger, S.; Kurts, C.; Heikenwälder, M.; Knolle, P.A.; Wohlleber, D. Transfer of MHC-class-I molecules among liver sinusoidal cells facilitates hepatic immune surveillance. J. Hepatol. 2014, 61, 600–608. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Croy, J.E.; Schirtzinger, L.; Torgerson, S.; Breyer, M.; Wroblewski, V.J. Aberrant bispecific antibody phar-macokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. MAbs 2016, 8, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Yao, Z.; Cheplowitz, A.M.; Suer, O.; Phillips, G.S.; Kwiek, J.; Rajaram, M.; Kim, J.; Robinson, J.M.; Ganesan, L.P.; et al. Mouse Liver Sinusoidal Endothelium Eliminates HIV-Like Particles from Blood at a Rate of 100 Million per Minute by a Second-Order Kinetic Process. Front. Immunol. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Anania, J.C.; Chenoweth, A.M.; Wines, B.D.; Hogarth, P.M. The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease. Front. Immunol. 2019, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kometani, K.; Minato, N.; Hamazaki, Y. Bone Marrow Endothelial Cells Take Up Blood-Borne Immune Complexes via Fcγ Receptor IIb2 in an Erythropoietin-Dependent Manner. J. Immunol. 2020, 205, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.K.; Liu, J.C.; Innes, B.T.; MacParland, S.A.; McGilvray, I.D.; Bader, G.D.; Keller, G.M. Generation of Functional Liver Sinusoidal Endothelial Cells from Human Pluripotent Stem-Cell-Derived Venous Angioblasts. Cell Stem Cell 2020, 27, 254–269.e9. [Google Scholar] [CrossRef]
- Patel, K.R.; Roberts, J.T.; Barb, A.W. Multiple variables at the leukocyte cell surface Impact Fcγ receptor-dependent mecha-nisms. Front. Immunol. 2019, 10, 223. [Google Scholar] [CrossRef]
- Bhandari, S.; Larsen, A.K.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. The Scavenger Function of Liver Sinusoidal Endothelial Cells in Health and Disease. Front. Physiol. 2021, 12, 1711. [Google Scholar] [CrossRef]
- Horst, A.K.; Kumashie, K.G.; Neumann, K.; Diehl, L.; Tiegs, G. Antigen presentation, autoantibody production, and therapeu-tic targets in autoimmune liver disease. Cell Mol. Immunol. 2020, 18, 92–111. [Google Scholar] [CrossRef] [PubMed]
- James, B.H.; Papakyriacou, P.; Gardener, M.J.; Gliddon, L.; Weston, C.J.; Lalor, P.F. The contribution of liver sinusoidal endo-thelial cells to clearance of therapeutic antibody. Front. Physiol. 2022, 12, 753833. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.L.; Mener, A.; Patel, S.R.; Jajosky, R.P.; Bennett, A.L.; Arthur, C.M.; Hendrickson, J.E.; Stowell, S.R. Antibody-mediated immune suppression by antigen modulation is antigen-specific. Blood Adv. 2018, 2, 2986–3000. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Leal, Y.; Marjoram, D.; Lazarus, A.H. Erythrocyte saturation with IgG is required for inducing antibody-mediated im-mune suppression and impacts both erythrocyte clearance and antigen-medulation mechanisms. J. Immunol. 2018, 200, 1295–1305. [Google Scholar] [CrossRef]
- Westhoff, C.M. AMIS and antigen modulation: Of mice and men. Blood 2016, 128, 3026–3028. [Google Scholar] [CrossRef][Green Version]
- Beum, P.V.; Kennedy, A.D.; Williams, M.E.; Lindorfer, M.A.; Taylor, R.P. The Shaving Reaction: Rituximab/CD20 Complexes Are Removed from Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Cells by THP-1 Monocytes. J. Immunol. 2006, 176, 2600–2609. [Google Scholar] [CrossRef]
- Daubeuf, S.; Lindorfer, M.A.; Taylor, R.P.; Joly, E.; Hudrisier, D. The direction of plasma membrane exchange between lym-phocytes and accessory cells by trogocytosis is influenced by the nature of the accessory cell. J. Immunol. 2010, 184, 1897–1908. [Google Scholar] [CrossRef]
- Crickx, E.; Chappert, P.; Sokal, A.; Weller, S.; Azzaoui, I.; Vandenberghe, A.; Bonnard, G.; Rossi, G.; Fadeev, T.; Storck, S.; et al. Rituximab-resistant splenic memory B cells and newly engaged naive B cells fuel relapses in patients with im-mune thrombocytopenia. Sci. Transl. Med. 2021, 13, eabc3961. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; Jones, J.D.; Hamilton, B.J.; Danilov, A.V.; Rigby, W.F.C. Role for ZAP-70 Signaling in the Differential Effector Functions of Rituximab and Obinutuzumab (GA101) in Chronic Lymphocytic Leukemia B Cells. J. Immunol. 2017, 199, 1275–1282. [Google Scholar] [CrossRef]
- Miot, S.; Marfurt, J.; Lach-Trifilieff, E.; Gonzalez-Rubio, C.; Lopez-Tracasa, M.; Sadallah, S.; Schifferli, J.A. The mechanism of loss of CR1 during maturation of erythrocytes is different between Factor I deficient patients and healthy donors. Blood Cells Mol. Dis. 2002, 29, 200–212. [Google Scholar] [CrossRef]
- Khera, R.; Das, N. Complement Receptor 1: Disease associations and therapeutic implications. Mol. Immunol. 2009, 46, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils kill antibody-opsonized cancer cells by trogocytosis. Cell Rep. 2018, 23, 3946–3959. [Google Scholar] [CrossRef]
- Valgardsdottir, R.; Cattaneo, I.; Klein, C.; Introna, M.; Figliuzzi, M.; Golay, J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood 2017, 129, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, R.; Challa, D.K.; Ram, S.; Ober, R.J.; Ward, E.S. Macrophage-mediated trogocytosis leads to death of antibody-opsonized tumor cells. Mol. Cancer Ther. 2016, 15, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Fries, L.F.; Siwik, S.A.; Malbran, A.; Frank, M.M. Phagocytosis of target particles bearing C3b-IgG covalent complexes by human monocytes and polymorphonuclear leukocytes. Immunology 1987, 62, 45–51. [Google Scholar]
- Ehlenberger, A.G.; Nussenzweig, V. The role of membrane receptors for C3b and C3d in phagocytosis. J. Exp. Med. 1977, 145, 357–371. [Google Scholar] [CrossRef]
- Schreiber, A.D.; Frank, M.M. Role of antibody and complement in the immune clearance and destruction of erythrocytes: I. In vivo effects of IgG and IgM complement fixing sites. J. Clin. Investig. 1972, 51, 575–582. [Google Scholar] [CrossRef]
- Schreiber, A.D.; Frank, M.M. Role of Antibody and Complement in the Immune Clearance and Destruction of Erythrocytes II. molecular nature of IgG and IgM complement-fixing sites and effects of their interaction with serum. J. Clin. Investig. 1972, 51, 583–589. [Google Scholar] [CrossRef]
- Atkinson, J.P.; Frank, M.M. Studies on the In Vivo Effects of Antibody interaction of IgM antibody and complement in the immune clearance and destruction of erythrocytes in man. J. Clin. Investig. 1974, 54, 339–348. [Google Scholar] [CrossRef]
- Atkinson, J.P.; Schreiber, A.D.; Frank, M.M. Effects of corticosteroids and splenectomy on the immune clearance and de-struction of erythrocytes. J. Clin. Investig. 1973, 52, 1509–1517. [Google Scholar] [CrossRef]
- Beurskens, F.J.; Lindorfer, M.A.; Farooqui, M.; Beum, P.V.; Engelberts, P.J.; Mackus, W.J.M.; Parren, P.; Wiestner, A.; Taylor, R.P. Exhaustion of Cytotoxic Effector Systems May Limit Monoclonal Antibody-Based Immunotherapy in Cancer Patients. J. Immunol. 2012, 188, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Densmore, J.J.; Pawluczkowycz, A.W.; Beum, P.V.; Kennedy, A.D.; Lindorfer, M.A.; Hamil, S.H.; Eggleton, J.C.; Taylor, R.P. Thrice-Weekly Low-Dose Rituximab Decreases CD20 Loss via Shaving and Promotes Enhanced Targeting in Chronic Lymphocytic Leukemia. J. Immunol. 2006, 177, 7435–7443. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Analyses of CD20 monoclonal antibody-mediated tumor cell killing mechanisms: Rational de-sign of dosing strategies. Mol. Pharm. 2014, 86, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.L.; Montalvao, F.; Celli, S.; Michonneau, D.; Breart, B.; Garcia, Z.; Perro, M.; Freytag, O.; Gerdes, C.A.; Bousso, P. Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of gly-coengineered anti-CD20 antibodies. Nat. Sci. Rep. 2016, 6, 34382. [Google Scholar] [CrossRef] [PubMed]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20–mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef]
- Gul, N.; Babes, K.; Korthouwer, R.; Bogels, M.; Braster, R.; Vidarsson, G.; Hagen, T.L.M.t.; Kubes, P.; van Egmond, M. Macro-phages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef]
- Church, A.K.; VanDerMeid, K.R.; Baig, N.A.; Baran, A.M.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S. Anti-CD20 monoclo-nal antibody-dependent phagocytosis of chronic lymphocytic leukaemia cells by autologous macrophages. Clin. Exp. Immunol. 2016, 183, 90–101. [Google Scholar] [CrossRef]
- Zent, C.S.; Elliott, M.R. Maxed out macs: Physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2016, 284, 1021–1039. [Google Scholar] [CrossRef]
- Pinney, J.J.; Rivera-Escalera, F.; Chu, C.C.; Whitehead, H.E.; vanDerMeid, K.R.; Nelson, A.M.; Barbeau, M.C.; Zent, C.S.; Elliott, M.R. Macrophage hypophagia as a mechanism of innate immune exhaustion in mAb-induced cell clearance. Blood 2020, 136, 2065–2079. [Google Scholar] [CrossRef]
- McBride, H.J.; Jassum, S.; Chow, V.; Kanakaraj, P.; Lebrec, H.; Kuhns, S.; Ferbas, J.; Wong, M.; Thway, T.M. Non-clinical similarity of biosimilar ABP 798 with rituximab reference product. Biologicals 2021, 72, 42–53. [Google Scholar] [CrossRef]
- Zent, C.S.; Wang, X.V.; Ketterling, R.; Hanson, C.A.; Libby, E.N.; Barrientos, J.C.; Call, T.G.; Chang, E.; Liu, J.J.; Calvo, A.R.; et al. A phase II randomized trial comparing standard and low dose rituximab combined with alemtuzumab as initial treatment of progressive chronic lymphocytic leukemia in older patients: A trial of the ECOG-ACRIN cancer research group (E1908). Am. J. Hematol. 2015, 91, 308–312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zent, C.S.; Taylor, R.P.; Lindorfer, M.A.; Beum, P.V.; LaPlant, B.; Wu, W.; Call, T.G.; Bowen, D.A.; Conte, M.J.; Frederick, L.A.; et al. Chemoimmunotherapy for relapsed/refractory and progressive 17p13 deleted chronic lymphocytic leukemia (CLL) combining pentostatin, alemtuzumab, and low dose rituximab is effective and tolerable and limits loss of CD20 expres-sion by circulating CLL cells. Am. J. Hematol. 2014, 89, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Leal, Y.; Lazarus, A.H. Could antigen loss be a potential mechanism to explain antibody-mediated immune suppres-sion? Transfusion 2021, 61, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, H.C.; Arthur, C.M.; Thompson, L.; Patel, S.R.; Stowell, S.R.; Hendrickson, J.E.; Lazarus, A.H. Anti-RhD reduces levels of detectable RhD antigen following anti-RhD infusion. Transfusion 2018, 58, 542–544. [Google Scholar] [CrossRef]
- Liu, J.; Santhanakrishnan, M.; Natarajan, P.; Gibb, D.R.; Eisenbarth, S.C.; Tormey, C.A.; Siddon, A.J.; Stowell, S.R.; Branch, D.R.; Hendrickson, J.E. Antigen modulation as a potential mechanism of anti-KEL immunoprophylaxis in mice. Blood 2016, 128, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab Infusion Promotes Rapid Complement Depletion and Acute CD20 Loss in Chronic Lymphocytic Leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar] [CrossRef]
- Oostindie, S.C.; Van Der Horst, H.J.; Lindorfer, M.A.; Cook, E.M.; Tupitza, J.C.; Zent, C.S.; Burack, R.; VanDerMeid, K.R.; Strumane, K.; Chamuleau, M.E.D.; et al. CD20 and CD37 antibodies synergize to activate complement by Fc-mediated clustering. Haematologica 2019, 104, 1841–1852. [Google Scholar] [CrossRef]
- Williams, E.L.; Tutt, A.L.; Beers, S.A.; French, R.R.; Chan, C.H.; Cox, K.L.; Roghanian, A.; Penfold, C.A.; Butts, C.L.; Boross, P.; et al. Immunotherapy targeting inhibitory Fcγ receptor IIB (CD32b) in the mouse is limited by monoclonal antibody consumption and receptor internalization. J. Immunol. 2013, 191, 4130–4140. [Google Scholar] [CrossRef]
- Elayeb, R.; Tamagne, M.; Pinheiro, M.; Ripa, J.; Djoudi, R.; Bierling, P.; Pirenne, F.; Vingert, B. Anti-CD20 Antibody Prevents Red Blood Cell Alloimmunization in a Mouse Model. J. Immunol. 2017, 199, 3771–3780. [Google Scholar] [CrossRef]
- Sullivan, H.C.; Gerner-Smidt, C.; Nooka, A.K.; Arthur, C.M.; Thompson, L.; Mener, A.; Patel, S.R.; Yee, M.; Fasano, R.M.; Josephson, C.D.; et al. Daratumumab (anti-CD38) induces loss of CD38 on red blood cells. Blood 2017, 129, 3033–3037. [Google Scholar] [CrossRef]
- Alari-Pahissa, E.; Ataya, M.; Moraitis, I.; Campos-Ruiz, M.; Altadill, M.; Muntasell, A.; Moles, A.; Lopez-Botet, M. NK cells eliminate Epstein-Barr virus bound to B cells through a specific antibody-mediated uptake. PLoS Pathog. 2021, 17, e1009868. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Korde, N.; Kotecha, R.; Reger, R.; Bor, S.; Kazandjian, D.; Landgren, O.; Childs, R.W. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with my-eloma. Clin. Cancer Res. 2016, 22, 5211–5222. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D. Antibody-Drug Conjugates of Calicheamicin Derivative: Gemtuzumab Ozogamicin and Inotuzumab Ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.I.; Crowther, C.; Mkhize, N.N.; Morris, L. Measuring the ability of HIV-specific antibodies to mediate trogocy-tosis. J. Immunol. Methods 2018, 463, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; van de Donk, N.W.C.J.; van Bueren, J.L.; Ahmadi, T.; Sasser, A.K.; Jansson, R.K.; Kokhorts, H.M.; Parren, P.W. Discovery, development and mechanisms of action of the human CD38 antibody daratumumab. In Successful Drug Discovery; Fischer, J., Klein, C., Childers, W.E., Eds.; Chapter 7; Wiley: Hoboken, NJ, USA, 2018; Volume 3, pp. 153–195. [Google Scholar]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.; Zweegman, S.; van Meerloo, J.; Musters, R.J.; et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Minarik, J.; Novak, M.; Flodr, P.; Balcarkova, J.; Mlynarcikova, M.; Krhovska, P.; Pika, T.; Pikalova, Z.; Bacovsky, J.; Scudla, V. CD38-negative relapse in multiple myeloma after daratumumab-based chemotherapy. Eur. J. Haematol. 2017, 99, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- de Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef]
- Jabbour, E.; Paul, S.; Kantarjian, H. The clinical development of antibody–drug conjugates—Lessons from leukaemia. Nat. Rev. Clin. Oncol. 2021, 18, 418–433. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Van Der Velden, V.H.J.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; Van Dongen, J.J.M. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [PubMed]
- Guffroy, M.; Falahatpisheh, H.; Biddle, K.; Kreeger, J.; Obert, L.; Walters, K.; Goldstein, R.; Boucher, G.; Coskran, T.; Reagan, W.; et al. Liver Microvascular Injury and Thrombocytopenia of Antibody–Calicheamicin Conjugates in Cynomolgus Monkeys—Mechanism and Monitoring. Clin. Cancer Res. 2017, 23, 1760–1770. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- McDonald, G.B.; Freston, J.W.; Boyer, J.L.; Deleve, L.D. Liver Complications Following Treatment of Hematologic Malignancy With Anti-CD22-Calicheamicin (Inotuzumab Ozogamicin). Hepatology 2018, 69, 831–844. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implica-tions. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Suzuki, E.; Kataoka, T.R.; Hirata, M.; Kawaguchi, K.; Nishie, M.; Haga, H.; Toi, M. Trogocytosis-mediated expression of HER2 on immune cells may be associated with a pathological complete response to trastuzumab-based primary systemic therapy in HER2-overexpressing breast cancer patients. BMC Cancer 2015, 15, 39. [Google Scholar] [CrossRef]
- Golay, J.; Lazzari, M.; Facchinetti, V.; Bernasconi, S.; Borleri, G.; Barbui, T.; Rambaldi, A.; Introna, M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: Further regulation by CD55 and CD59. Blood 2001, 98, 3383–3389. [Google Scholar] [CrossRef]
- Glennie, M.J.; French, R.R.; Cragg, M.S.; Taylor, R.P. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 2007, 44, 3823–3837. [Google Scholar] [CrossRef]
- van Rees, D.J.; Brinkhaus, M.; Klein, B.; Verkuijlen, P.; Tool, A.T.; Schornagel, K.; Treffers, L.W.; van Houdt, M.; Kater, A.P.; Vidarsson, G.; et al. Sodium stibogluconate and CD47-SIRPα blockade overcome resistance of anti-CD20-opsonized B cells to neutrophil killing. Blood Adv. 2022, 6, 2156–2166. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Lipfert, L.; Chevalier, K.; Bushey, B.S.; Henley, B.; Lenhart, R.; Sendecki, J.; Beqiri, M.; Millar, H.J.; Packman, K.; et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/cMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol. Cancer Ther. 2020, 19, 2044–2056. [Google Scholar] [CrossRef]
- Liew, P.X.; Kim, J.H.; Lee, W.-Y.; Kubes, P. Antibody-dependent fragmentation is a newly identified mechanism of cell killing in vivo. Sci. Rep. 2017, 7, 10515. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Jansen, J.H.M.; Pastula, A.; van der Poel, C.E.; Leusen, J.H.W. Both activating and inhibitory Fcγ receptors mediate rituximab-induced trogocytosis of CD20 in mice. Immunol. Lett. 2012, 143, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Rowley, T.F.; Junker, F.; Peters, S.J.; Crilly, S.; Compson, J.; Eddleston, A.; Björkelund, H.; Greenslade, K.; Parkinson, M.; et al. Multivalent Fcγ-receptor engagement by a hexameric Fc-fusion protein triggers Fcγ-receptor internalisation and modulation of Fcγ-receptor functions. Sci. Rep. 2017, 7, 17049. [Google Scholar] [CrossRef]
- Fitzpatrick, E.A.; Wang, J.; Strome, S.E. Engineering of Fc Multimers as a Protein Therapy for Autoimmune Disease. Front. Immunol. 2020, 11, 496. [Google Scholar] [CrossRef]
- Repik, A.; Pincus, S.E.; Ghiran, I.; Nicholson-Weller, A.; Asher, D.R.; Cerny, A.M.; Casey, L.S.; Jones, S.M.; Jones, S.N.; Mohamed, N.; et al. A transgenic mouse model for studying the clearance of blood-borne pathogens via human complement recep-tor 1 (CR1). Clin. Exp. Immunol. 2005, 140, 230–240. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Hahn, C.S.; Foley, P.L.; Taylor, R.P. Heteropolymer-mediated clearance of immune complexes via erythro-cyte CR1: Mechanisms and applications. Immunol. Rev. 2001, 183, 10–24. [Google Scholar] [CrossRef]
- Beers, S.A.; French, R.R.; Chan, H.T.C.; Lim, S.H.; Jarrett, T.C.; Vidal, R.M.; Wijayaweera, S.S.; Dixon, S.V.; Kim, H.; Cox, K.L.; et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: Implications for antibody selection. Blood 2010, 115, 5191–5201. [Google Scholar] [CrossRef]
- Roghanian, A.; Teige, I.; Mårtensson, L.; Cox, K.L.; Kovacek, M.; Ljungars, A.; Mattson, J.; Sundberg, A.; Vaughan, A.T.; Shah, V.; et al. Antagonistic human FcgRIIB (CD32B) antibodies have anti-tumour activity and overcome resistance to antibody therapy in vivo. Cancer Cell 2015, 27, 473–488. [Google Scholar] [CrossRef]
- Beum, P.V.; Peek, E.M.; Lindorfer, M.A.; Beurskens, F.J.; Engelberts, P.J.; Parren, P.W.H.I.; Van De Winkel, J.G.J.; Taylor, R.P.; Labrijn, A.F.; Meesters, J.; et al. Loss of CD20 and Bound CD20 Antibody from Opsonized B Cells Occurs More Rapidly Because of Trogocytosis Mediated by Fc Receptor-Expressing Effector Cells Than Direct Internalization by the B Cells. J. Immunol. 2011, 187, 3438–3447. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.T. Three major uncertainties in the antibody therapy of cancer. Haematologica 2014, 99, 1538–1546. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindorfer, M.A.; Taylor, R.P. FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis. Antibodies 2022, 11, 45. https://doi.org/10.3390/antib11030045
Lindorfer MA, Taylor RP. FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis. Antibodies. 2022; 11(3):45. https://doi.org/10.3390/antib11030045
Chicago/Turabian StyleLindorfer, Margaret A., and Ronald P. Taylor. 2022. "FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis" Antibodies 11, no. 3: 45. https://doi.org/10.3390/antib11030045
APA StyleLindorfer, M. A., & Taylor, R. P. (2022). FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis. Antibodies, 11(3), 45. https://doi.org/10.3390/antib11030045