
Generation of Antibodies Selectively Recognizing Epitopes in a Formaldehyde-Fixed Cell-Surface Antigen Using Virus-like Particle Display and Hybridoma Technology

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cells
2.3. Establishment of VLP Producer Cells and Murine Cells Expressing Human trNGFR
2.4. VLP Preparation
2.5. Fixation of Cells and VLPs
2.6. Flow Cytometric Analysis during Fixation Protocol and Screening Cell Line Development
2.7. Protein Quantitation
2.8. VLP Capture Assay
2.9. Dynamic Light Scattering
2.10. Transmission Electron Microscopic Analysis
2.11. Statistical Analysis
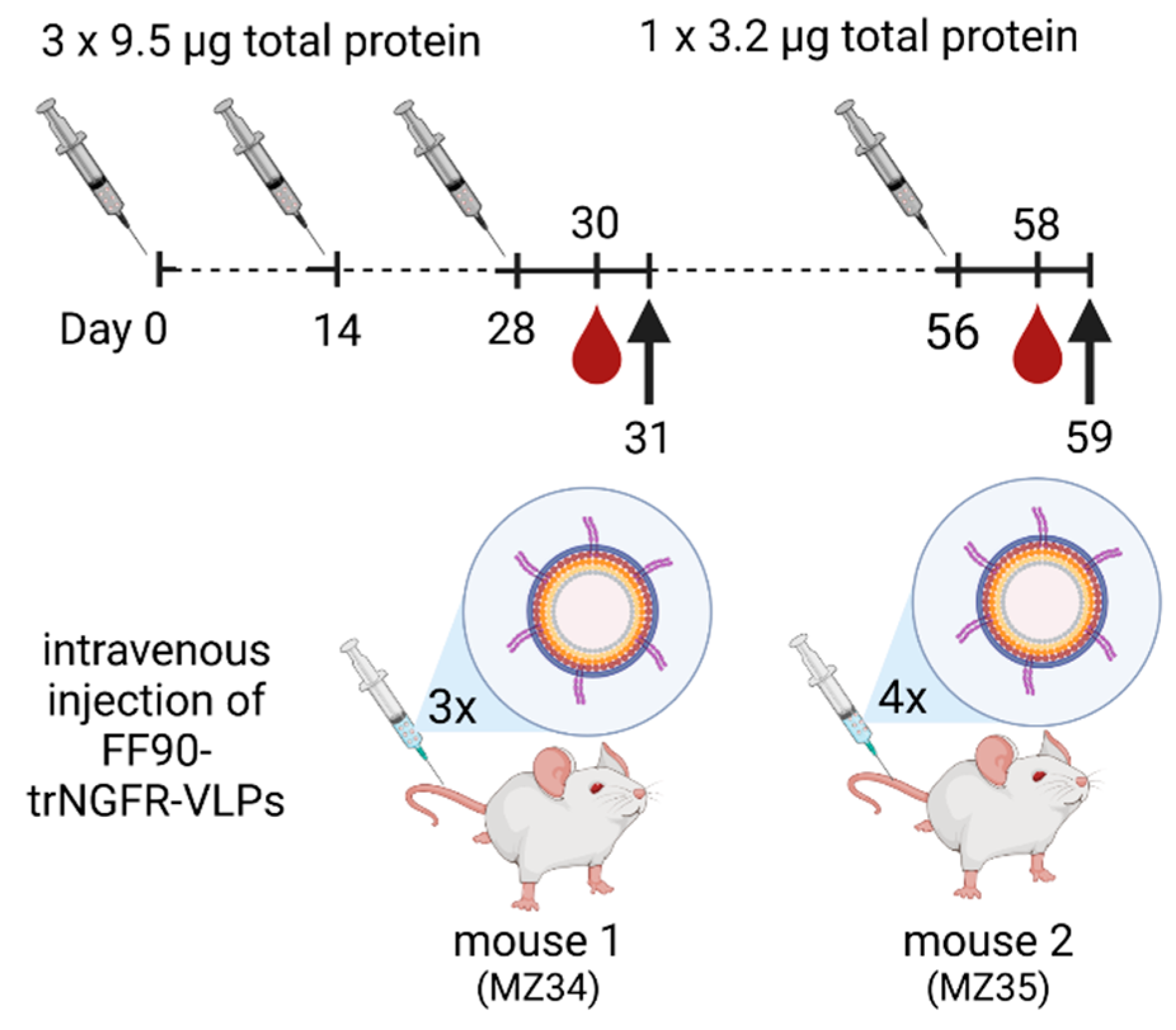
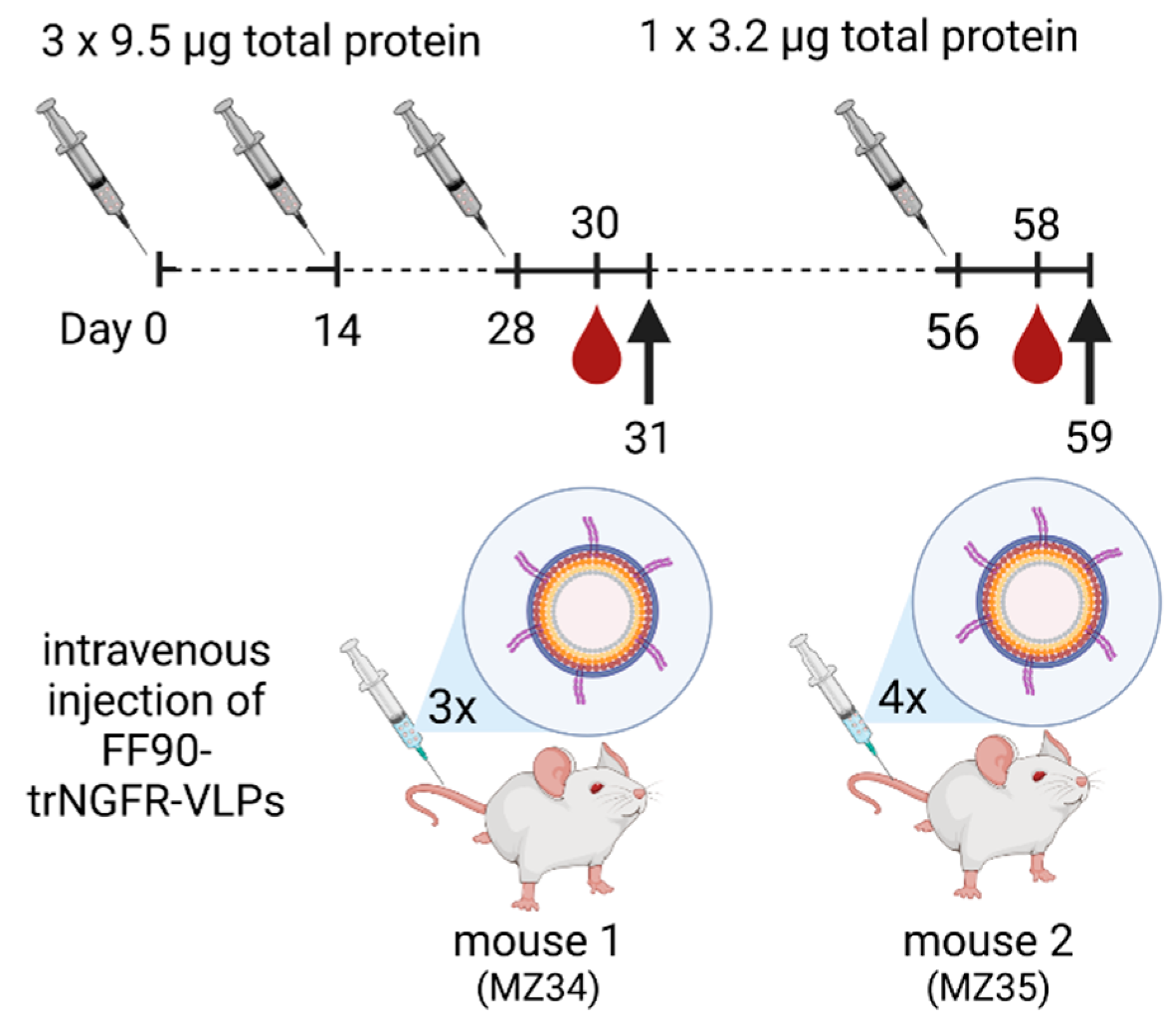
2.12. Immunization of Mice
2.13. Generation of Hybridoma Cells
2.14. Screening for Antibodies
3. Results and Discussion
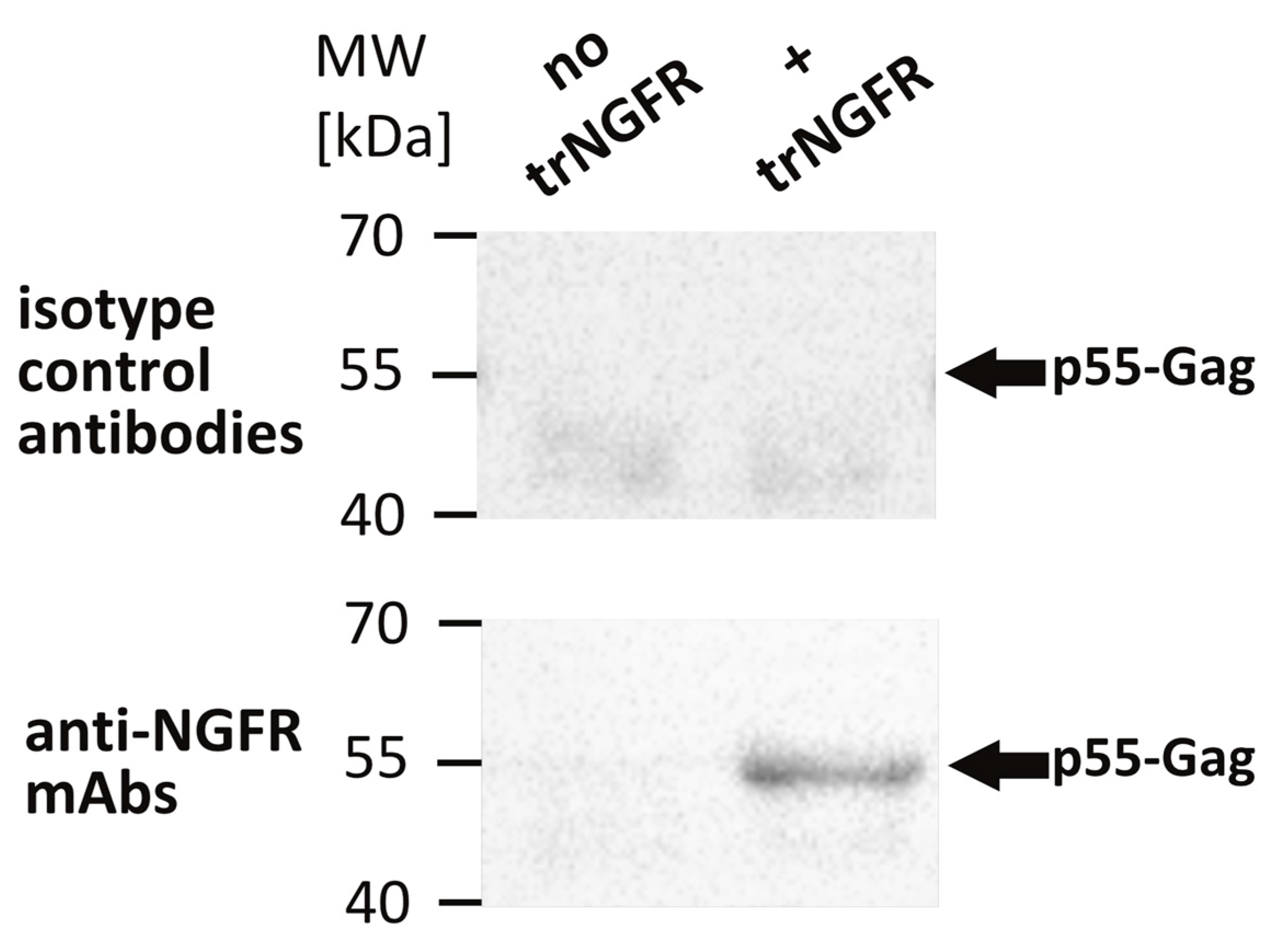
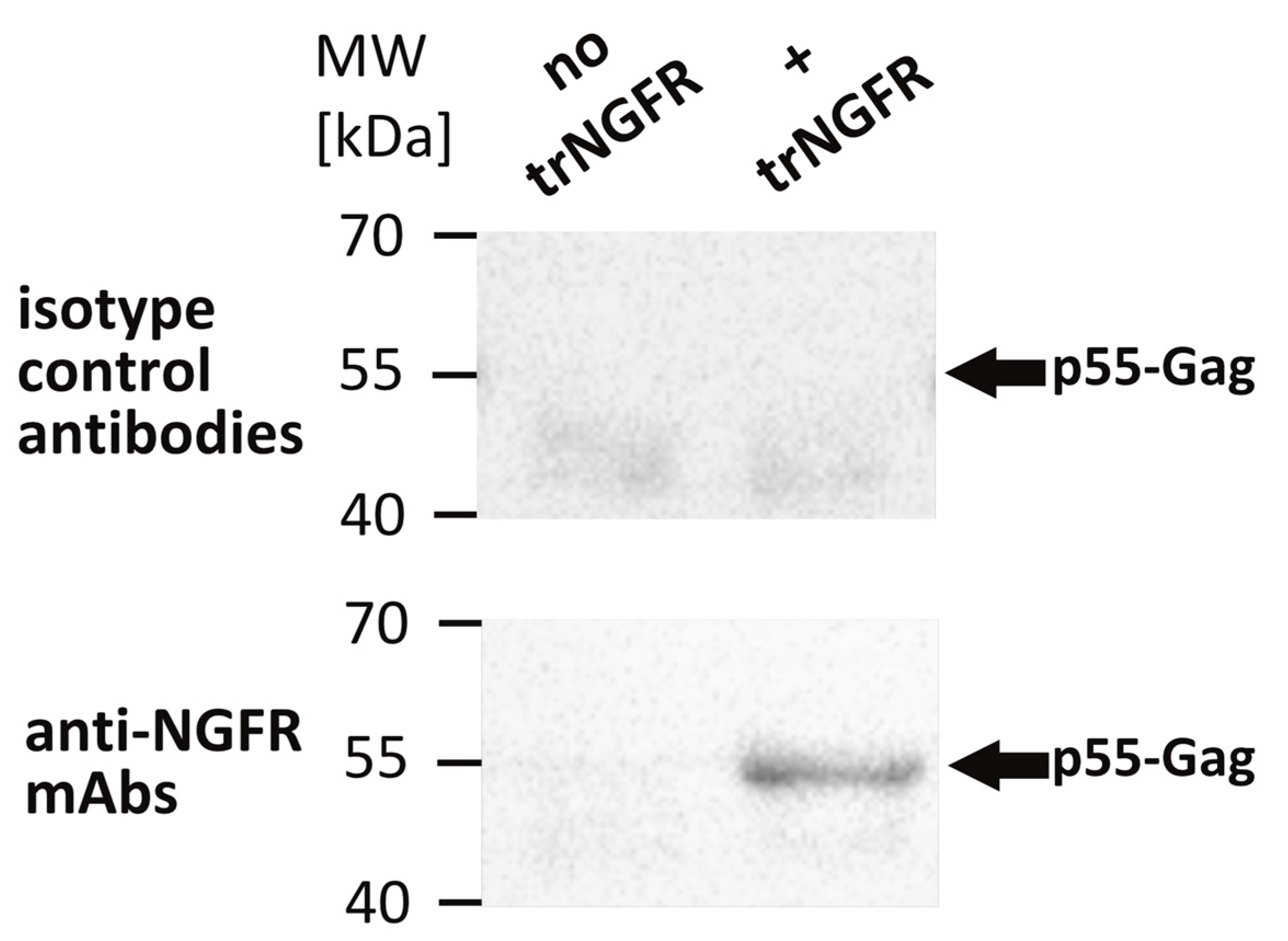
3.1. Characterization of VLP Producer Cell Pools
3.2. Development of a Fixation Protocol for Antigen Preparation
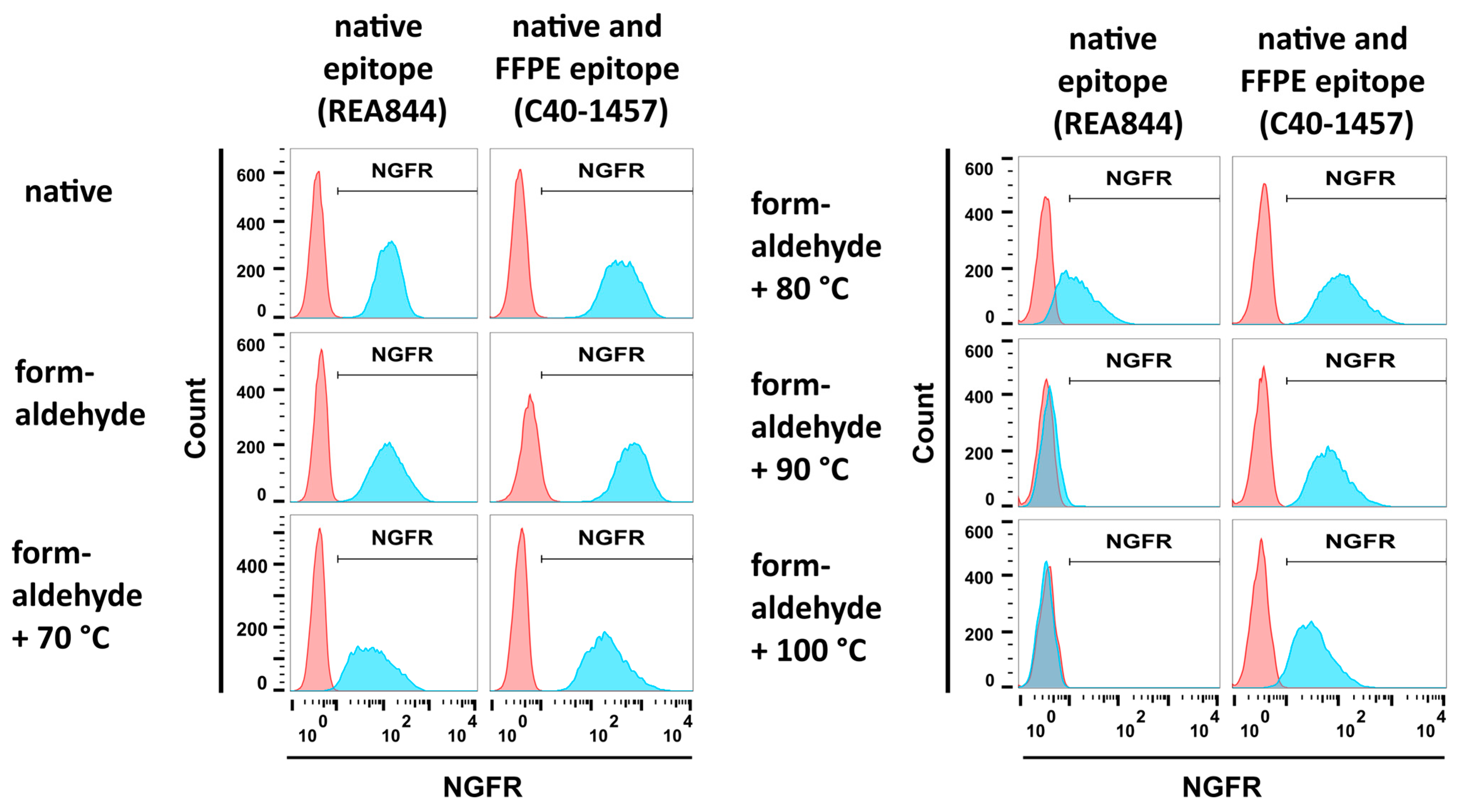
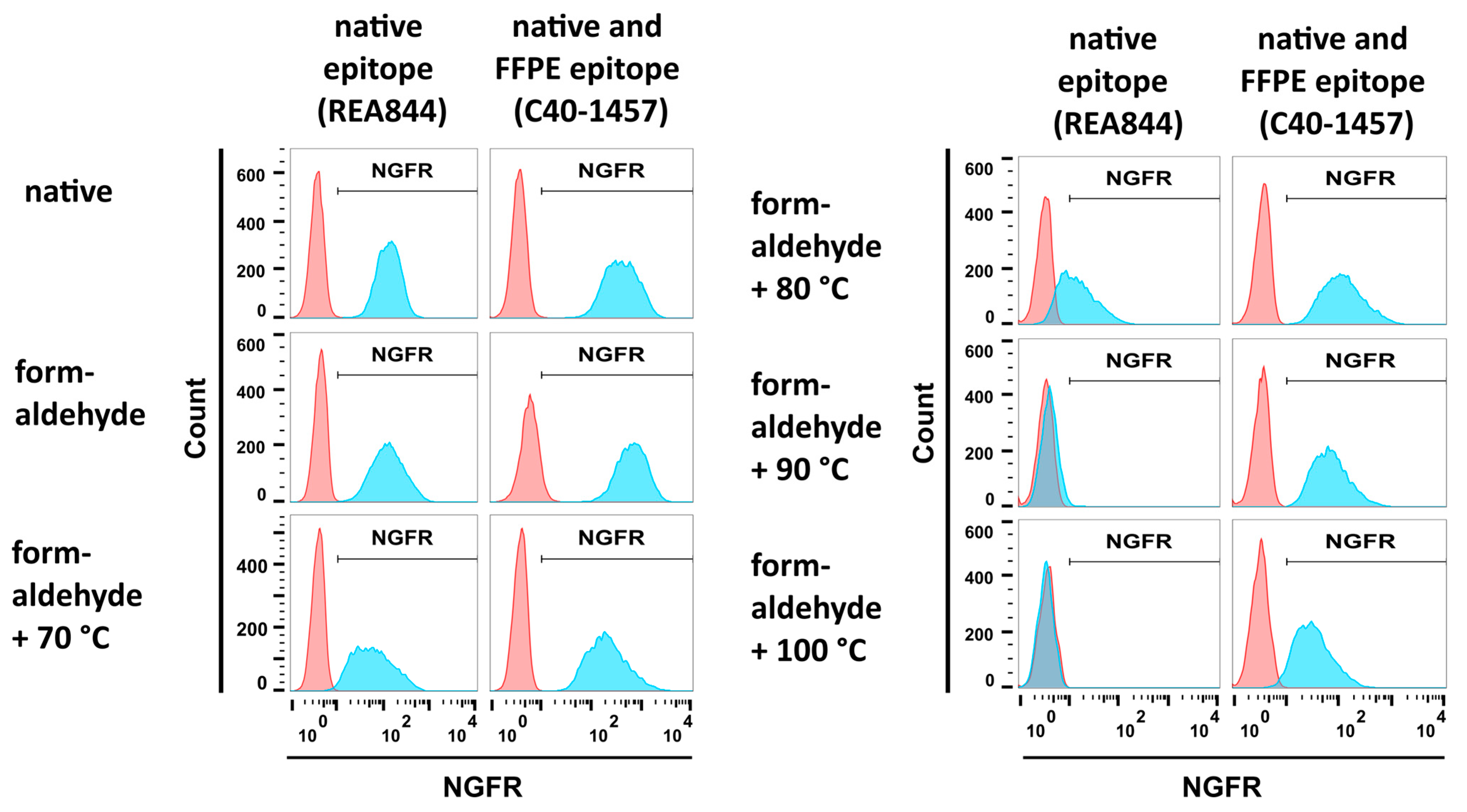
3.2.1. Formaldehyde and Heat Fixation induce a Loss of Epitope Recognition of an Antibody Directed against Native NGFR but Maintain Binding of an FFPE-Compatible Antibody
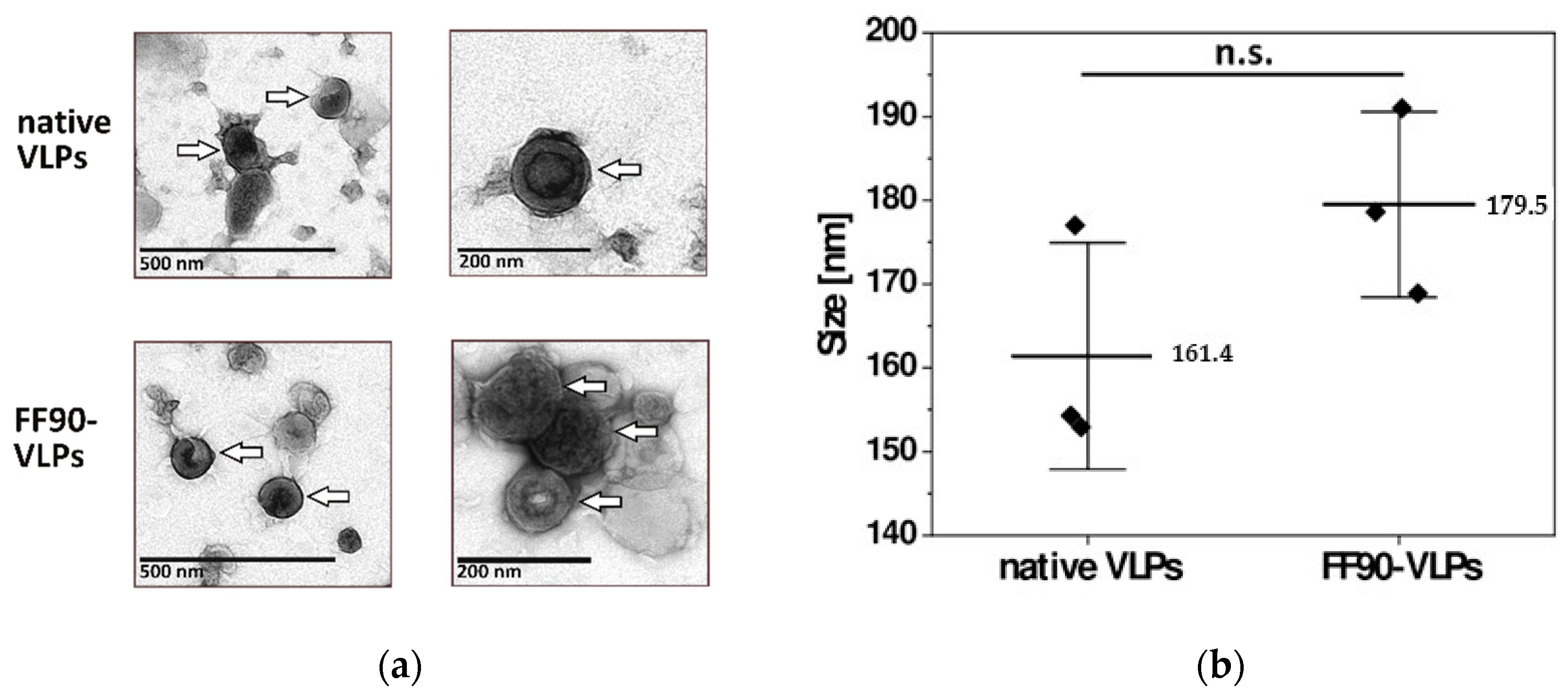
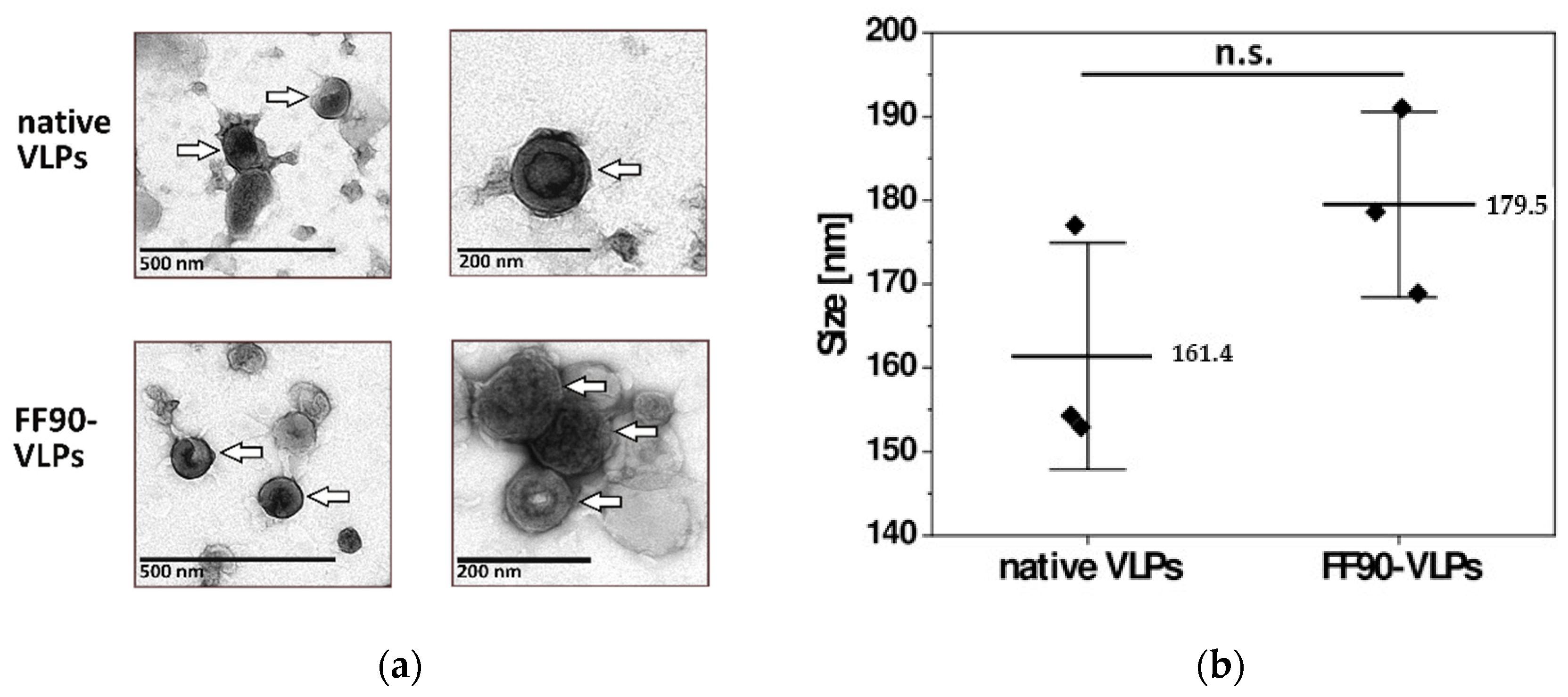
3.2.2. VLPs Maintain Their Integrity and Morphology after FF90-Treatment
3.3. Immunization of Mice, Generation of Hybridomas and Screening of Monoclonal Antibodies
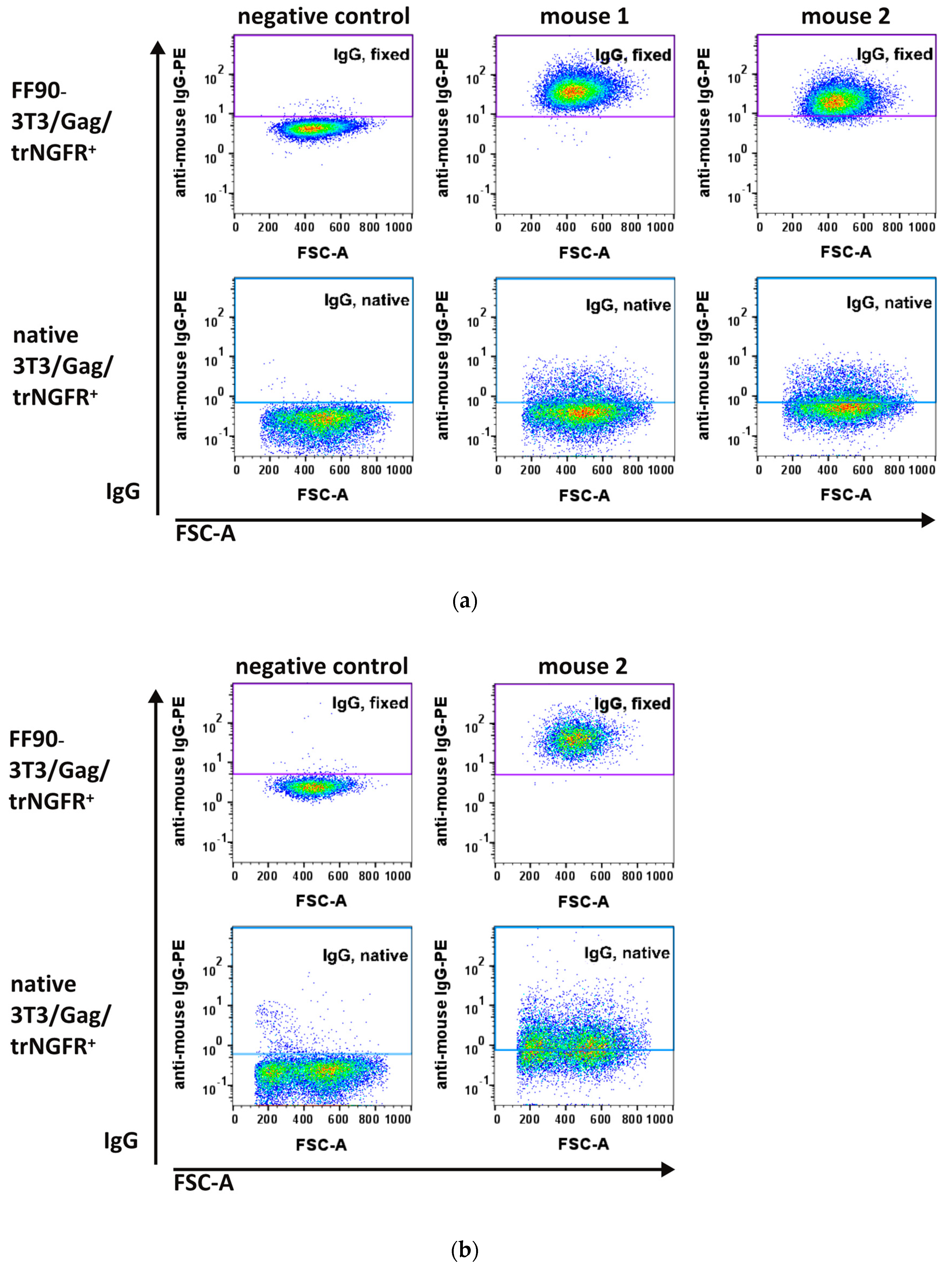
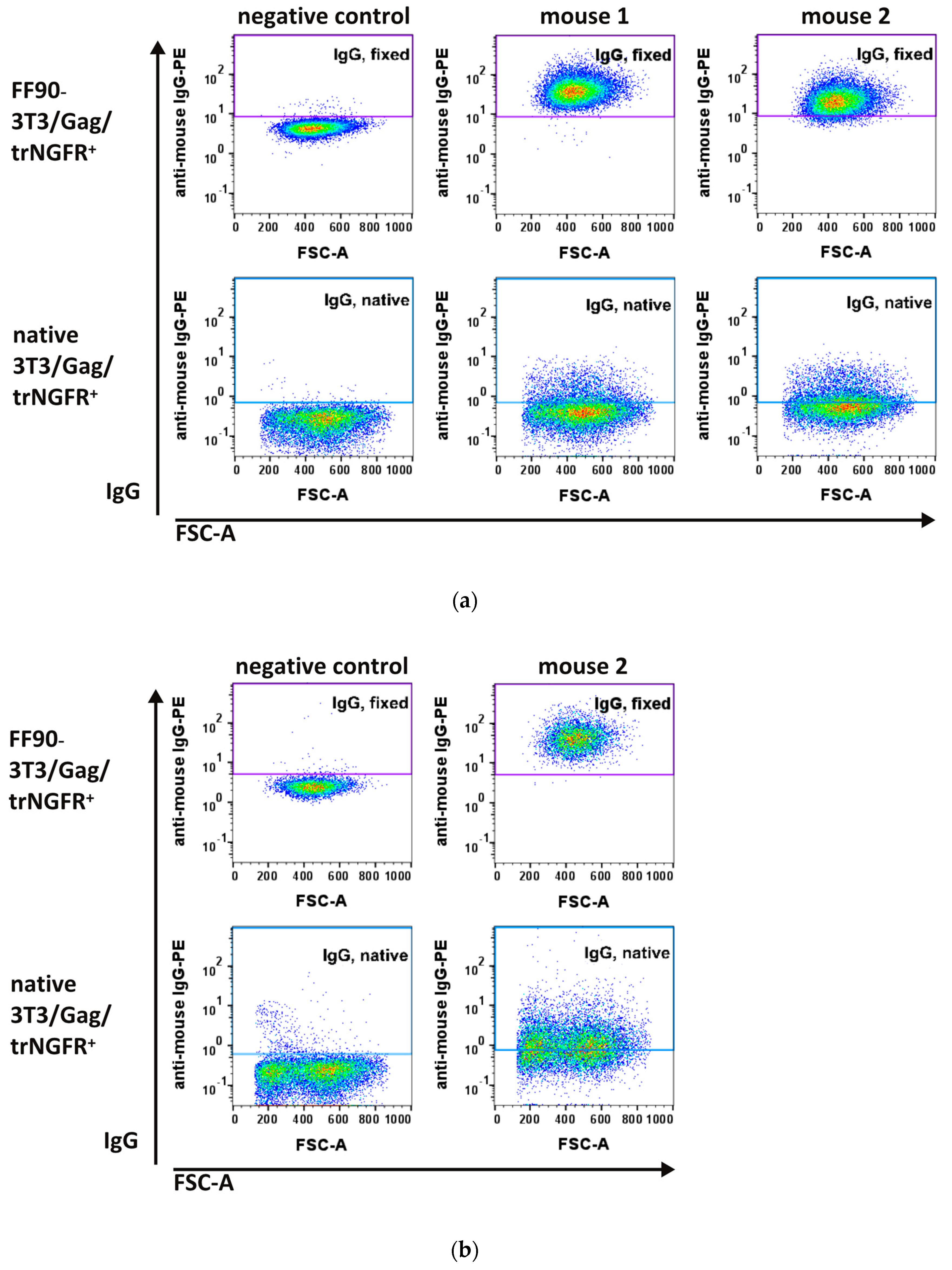
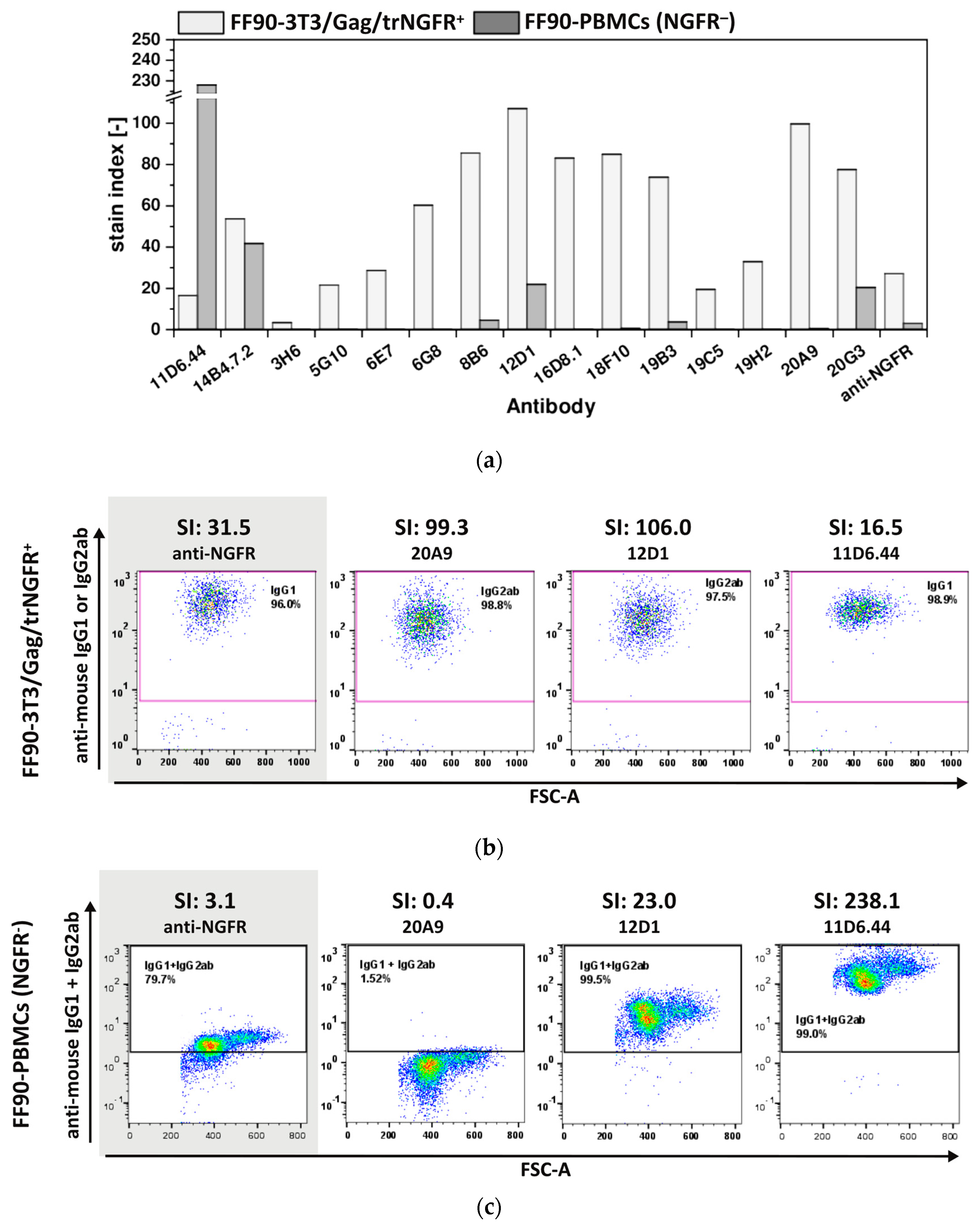
3.3.1. FF90-trNGFR-VLP Immunization Elicits IgG Antibodies Recognizing FF90-3T3/Gag/trNGFR Cells
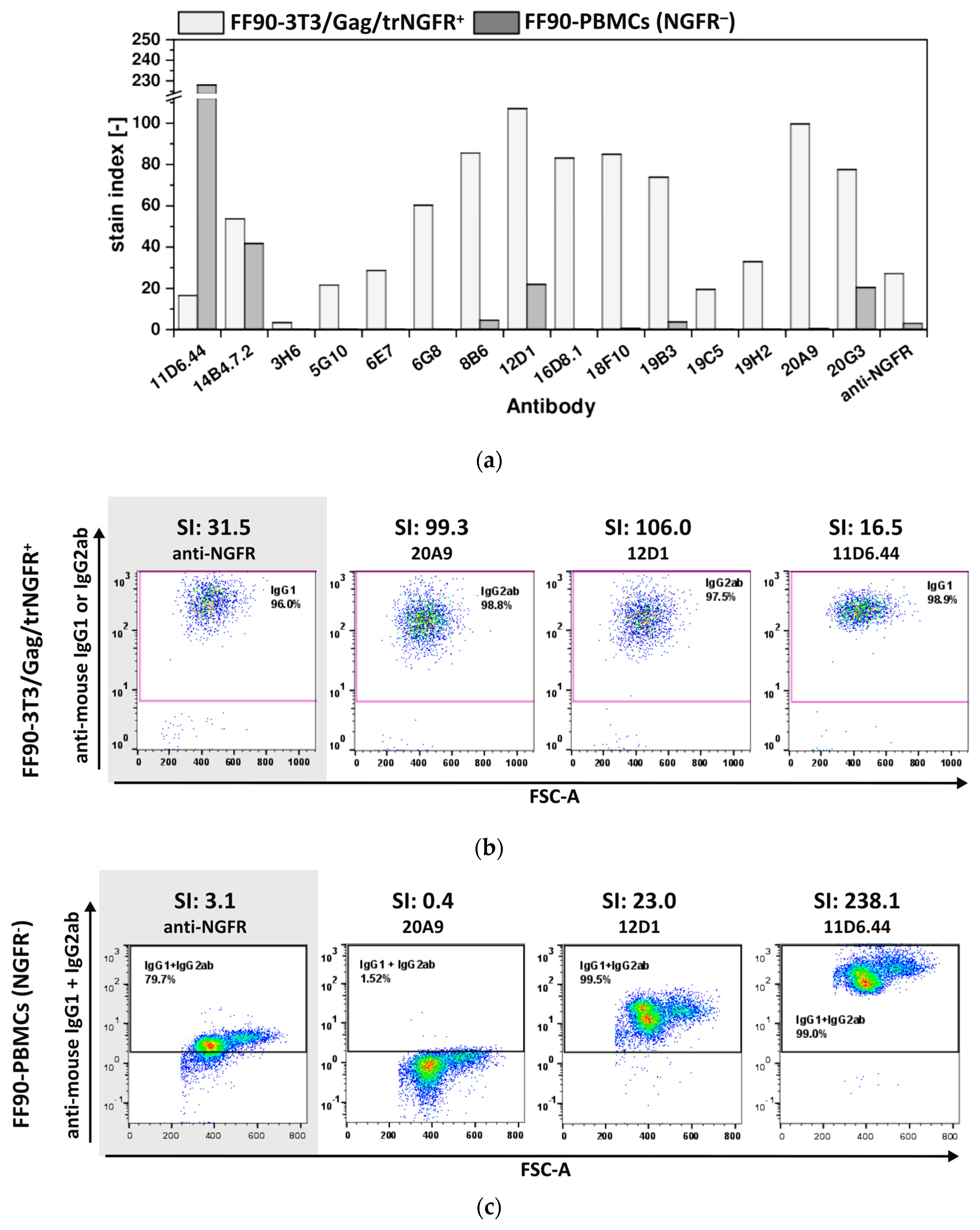
3.3.2. Hybridoma Cells Produce Antibodies Recognizing FF90-3T3/Gag/trNGFR Cells
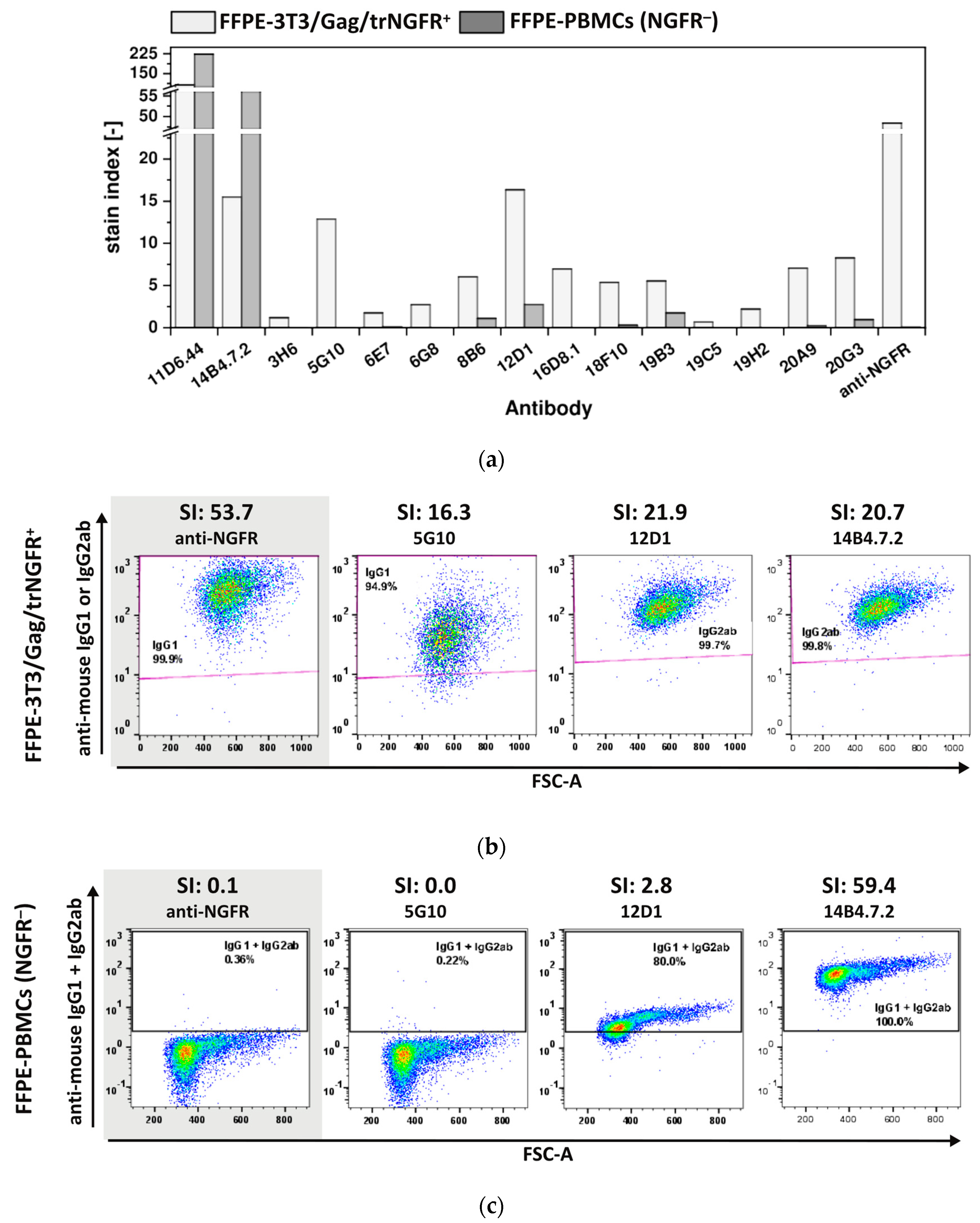
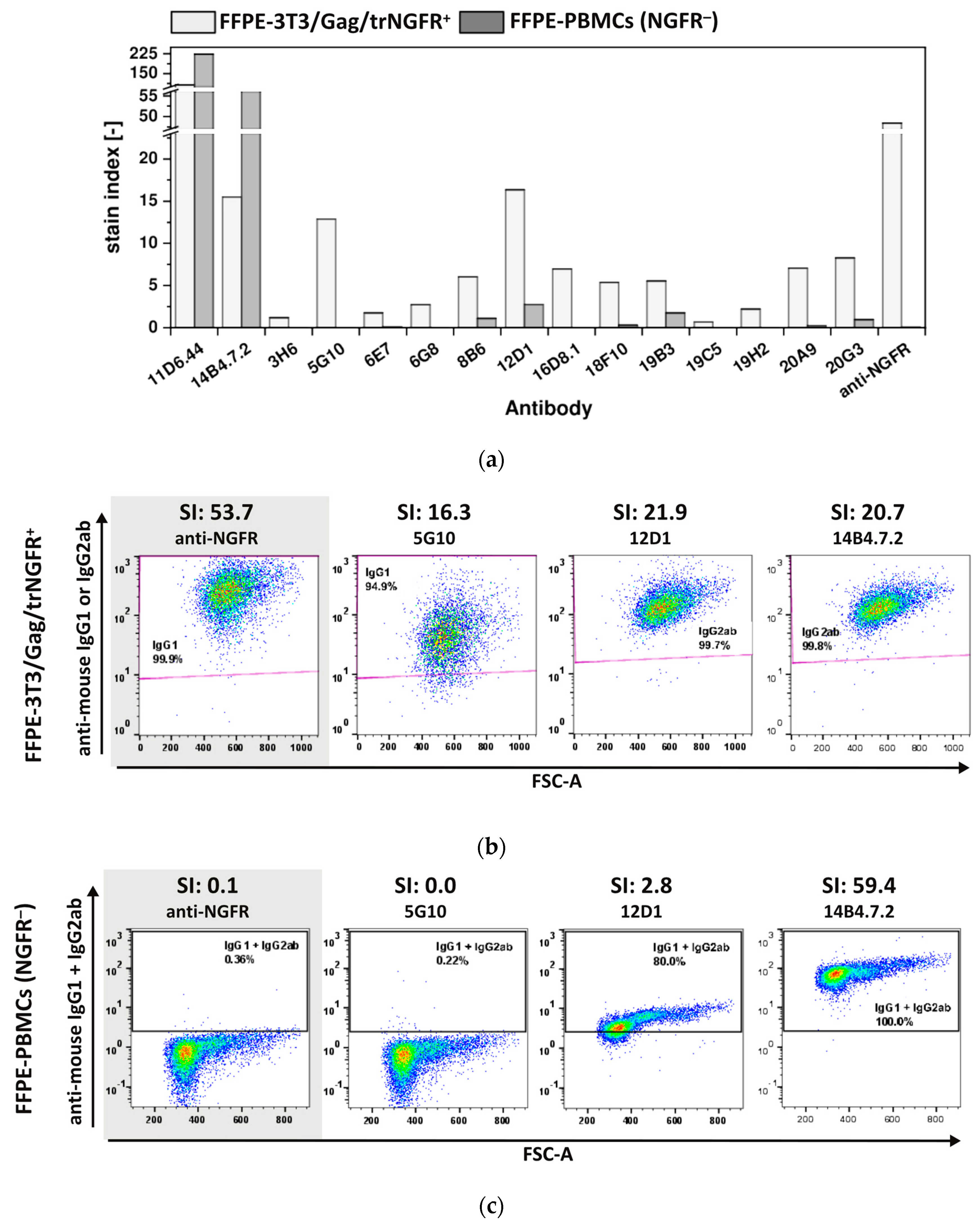
3.3.3. The Majority of FF90-NGFR Generated mAbs Enables Also the Detection of FFPE-NGFR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Meneses-Acosta, A.; Gómez, A.; Ramírez, O.T. Control of redox potential in hybridoma cultures: Effects on MAb production, metabolism, and apoptosis. J. Ind. Microbiol. Biotechnol. 2012, 39, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, E.F.; Riegman, P.H.; Grizzle, W.E.; Watson, P.H. Factors that drive the increasing use of FFPE tissue in basic and translational cancer research. Biotech. Histochem. Off. Public Biol. Stain. Comm. 2018, 93, 373–386. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A.; Miller, M.A. When tissue antigens and antibodies get along: Revisiting the technical aspects of immunohistochemistry-the red, brown, and blue technique. Vet. Pathol. 2014, 51, 42–87. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, A.-K.; Bosch, B.J.; Spitzbarth, I.; Lehmbecker, A.; Te, N.; Bensaid, A.; Segalés, J.; Baumgärtner, W. Detection of MERS-CoV antigen on formalin-fixed paraffin-embedded nasal tissue of alpacas by immunohistochemistry using human monoclonal antibodies directed against different epitopes of the spike protein. Vet. Immunol. Immunopathol. 2019, 218, 109939. [Google Scholar] [CrossRef]
- Werner, M.; Chott, A.; Fabiano, A.; Battifora, H. Effect of formalin tissue fixation and processing on immunohistochemistry. Am. J. Surg. Pathol. 2000, 24, 1016–1019. [Google Scholar] [CrossRef]
- Thavarajah, R.; Mudimbaimannar, V.K.; Elizabeth, J.; Rao, U.K.; Ranganathan, K. Chemical and physical basics of routine formaldehyde fixation. J. Oral Maxillofac. Pathol. JOMFP 2012, 16, 400–405. [Google Scholar] [CrossRef]
- Soares, H.R.; Castro, R.; Tomás, H.A.; Rodrigues, A.F.; Gomes-Alves, P.; Bellier, B.; Klatzmann, D.; Carrondo, M.J.T.; Alves, P.M.; Coroadinha, A.S. Tetraspanins displayed in retrovirus-derived virus-like particles and their immunogenicity. Vaccine 2016, 34, 1634–1641. [Google Scholar] [CrossRef]
- Schneider, I.C.; Hartmann, J.; Braun, G.; Stitz, J.; Klamp, T.; Bihi, M.; Sahin, U.; Buchholz, C.J. Displaying Tetra-Membrane Spanning Claudins on Enveloped Virus-Like Particles for Cancer Immunotherapy. Biotechnol. J. 2018, 13, e1700345. [Google Scholar] [CrossRef]
- Sailaja, G.; Skountzou, I.; Quan, F.-S.; Compans, R.W.; Kang, S.-M. Human immunodeficiency virus-like particles activate multiple types of immune cells. Virology 2007, 362, 331–341. [Google Scholar] [CrossRef]
- McFall-Boegeman, H.; Huang, X. Mechanisms of cellular and humoral immunity through the lens of VLP-based vaccines. Expert Rev. Vaccines 2022, 21, 453–469. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef]
- Yusa, K.; Zhou, L.; Li, M.A.; Bradley, A.; Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, R.; Gschwendtberger, T.; Felski, C.; Jahn, C.; Langer, F.; Sterneckert, J.; Hermann, A.; Lühmann, J.; Steinemann, D.; Haase, A.; et al. A selectable all-in-one CRISPR prime editing piggyBac transposon allows for highly efficient gene editing in human cell lines. Sci. Rep. 2021, 11, 22154. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.G.; Zhang, X.; Torti, F.; Chen, S.Y. Anti-HIV type 1 activity of wild-type and functional defective RANTES intrakine in primary human lymphocytes. Hum. Gene Ther. 1998, 9, 2005–2018. [Google Scholar] [CrossRef]
- Castellino, S.M.; Kurtzberg, J.; Smith, C. Retroviral vector-mediated gene transfer into umbilical cord blood-derived megakaryocyte and platelet progenitors. Biol. Blood Marrow Transplant. 1999, 5, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.P.M.; van Manen, D.; Vellinga, J.; Wegmann, F.; Callendret, B.C.S.; Krarup, A.; Stitz, J. Synthetic Human Immunodeficiency Virus (HIV) Envelope Antigen, Vectors, and Compositions Thereof. U.S. Patent 10,369,214, 6 August 2019. [Google Scholar]
- Rosengarten, J.F.; Schatz, S.; Wolf, T.; Barbe, S.; Stitz, J. Components of a HIV-1 vaccine mediate virus-like particle (VLP)-formation and display of envelope proteins exposing broadly neutralizing epitopes. Virology 2022, 568, 41–48. [Google Scholar] [CrossRef]
- Berg, K.; Schäfer, V.N.; Tschorn, N.; Stitz, J. Advanced Establishment of Stable Recombinant Human Suspension Cell Lines Using Genotype-Phenotype Coupling Transposon Vectors. Methods Mol. Biol. 2020, 2070, 351–361. [Google Scholar] [CrossRef]
- Berg, K.; Schäfer, V.N.; Bartnicki, N.; Eggenschwiler, R.; Cantz, T.; Stitz, J. Rapid establishment of stable retroviral packaging cells and recombinant susceptible target cell lines employing novel transposon vectors derived from Sleeping Beauty. Virology 2019, 531, 40–47. [Google Scholar] [CrossRef]
- Sharma, N.; Hollensen, A.K.; Bak, R.O.; Staunstrup, N.H.; Schrøder, L.D.; Mikkelsen, J.G. The impact of cHS4 insulators on DNA transposon vector mobilization and silencing in retinal pigment epithelium cells. PLoS ONE 2012, 7, e48421. [Google Scholar] [CrossRef]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.; Meyers, A.E.; Williamson, A.-L.; Rybicki, E.P. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol. J. 2012, 9, 210. [Google Scholar] [CrossRef]
- Maecker, H.T.; Frey, T.; Nomura, L.E.; Trotter, J. Selecting fluorochrome conjugates for maximum sensitivity. Cytom. Part A 2004, 62A, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Lavado-García, J.; Jorge, I.; Boix-Besora, A.; Vázquez, J.; Gòdia, F.; Cervera, L. Characterization of HIV-1 virus-like particles and determination of Gag stoichiometry for different production platforms. Biotechnol. Bioeng. 2021, 118, 2660–2675. [Google Scholar] [CrossRef] [PubMed]
- Rosengarten, J.F.; Schatz, S.; Stitz, J. Detection of Neutralization-sensitive Epitopes in Antigens Displayed on Virus-Like Particle (VLP)-Based Vaccines Using a Capture Assay. J. Vis. Exp. 2022, 180, e63137. [Google Scholar] [CrossRef]
- Schatz, S.; van Dijk, F.H.; Dubiel, A.E.; Cantz, T.; Eggenschwiler, R.; Stitz, J. Generation of Human 293-F Suspension NGFR Knockout Cells Using CRISPR/Cas9 Coupled to Fluorescent Protein Expression. In Genotype Phenotype Coupling: Methods and Protocols; Zielonka, S., Krah, S., Eds.; Springer: New York, NY, USA, 2023; pp. 361–371. ISBN 978-1-0716-3279-6. [Google Scholar]
- Sharma, D.; Barhwal, K.K.; Biswal, S.N.; Srivastava, A.K.; Bhardwaj, P.; Kumar, A.; Chaurasia, O.P.; Hota, S.K. Hypoxia-mediated alteration in cholesterol oxidation and raft dynamics regulates BDNF signalling and neurodegeneration in hippocampus. J. Neurochem. 2019, 148, 238–251. [Google Scholar] [CrossRef]
- Jamali, A.; Kapitza, L.; Schaser, T.; Johnston, I.C.D.; Buchholz, C.J.; Hartmann, J. Highly Efficient and Selective CAR-Gene Transfer Using CD4- and CD8-Targeted Lentiviral Vectors. Mol. Ther.-Methods Clin. Dev. 2019, 13, 371–379. [Google Scholar] [CrossRef]
- Fujii, K.; Morita, S.; Mochizuki, M.; Shibuya-Takahashi, R.; Fujimori, H.; Yamaguchi, K.; Abe, J.; Yamazaki, T.; Imai, T.; Sugamura, K.; et al. Establishment of a monoclonal antibody against glycosylated CD271 specific for cancer cells in immunohistochemistry. Cancer Sci. 2022, 113, 2878–2887. [Google Scholar] [CrossRef]
- Lin, J.-R.; Izar, B.; Wang, S.; Yapp, C.; Mei, S.; Shah, P.M.; Santagata, S.; Sorger, P.K. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 2018, 7, e31657. [Google Scholar] [CrossRef]
- Johnston, I.C.D.; (Miltenyi Biotec, Bergisch Galdbach, Germany). List of anti-NGFR mAbs tested for FFPE tissue reactivity: The anti-NGFR mAb REA844 (Miltenyi Biotec, Germany) is not reactive with NGFR in FFPE tissue samples. Personal communication, 2021. [Google Scholar]
- Wright, E.R.; Schooler, J.B.; Ding, H.J.; Kieffer, C.; Fillmore, C.; Sundquist, W.I.; Jensen, G.J. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007, 26, 2218–2226. [Google Scholar] [CrossRef]
- Martin, J.L.; Cao, S.; Maldonado, J.O.; Zhang, W.; Mansky, L.M. Distinct Particle Morphologies Revealed through Comparative Parallel Analyses of Retrovirus-Like Particles. J. Virol. 2016, 90, 8074–8084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cubas, R.; Li, M.; Chen, C.; Yao, Q. Virus-like particle vaccine activates conventional B2 cells and promotes B cell differentiation to IgG2a producing plasma cells. Mol. Immunol. 2009, 46, 1988–2001. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, Y.-T.; Ko, E.-J.; Kim, K.-H.; Hwang, H.S.; Park, S.; Kwon, Y.-M.; Kang, S.M. Soluble F proteins exacerbate pulmonary histopathology after vaccination upon respiratory syncytial virus challenge but not when presented on virus-like particles. Hum. Vaccines Immunother. 2017, 13, 2594–2605. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | NGFR per 1 µg Gag | Number of Replicates |
|---|---|---|
| 293-F | <0.08 ng | n = 1 |
| 293-F/Mos1.Gag | <0.08 ng | n = 3 |
| 293-F/Mos1.Gag/trNGFR | 99.6 ± 8.5 ng | n = 3 |
| Immunization (ID) | Screen | Screening Cells | Binding mAbs/ Tested mAbs | IgG subclass |
|---|---|---|---|---|
| 3 x FF90-trNGFR-VLPs (MZ34) | 1st | native and FF90-3T3/Gag/trNGFR+ | 3/698 (0.4%) 1 | 1 × IgG1; 2 × IgG2ab |
| 2nd | FF90-PBMCs (NGFR⁻) | 2/2 (100%) 2 | ||
| 4 x FF90-trNGFR-VLPs (MZ35) | 1st | native and FF90-3T3/Gag/trNGFR+ | 35/975 (3.6%) 1 | 9 × IgG1; 26 × IgG2ab |
| 2nd | FF90-PBMCs (NGFR⁻) | 4/13 (30.8%) 2 |
| Immunization (ID) | Screening Cells | Binding mAbs/ Tested mAbs |
|---|---|---|
| 3 x FF90-trNGFR-VLPs (MZ34) | FFPE-3T3/Gag/trNGFR+ | 2/2 (100%) 1 |
| FFPE-PBMCs (NGFR−) | 2/2 (100%) 2 | |
| 4 x FF90-trNGFR-VLPs (MZ35) | FFPE-3T3/Gag/trNGFR+ | 12/13 (92.3%) 1 |
| FFPE-PBMCs (NGFR−) | 3/12 (25.0%) 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schatz, S.; Willnow, L.; Winkels, M.; Rosengarten, J.F.; Theek, B.; Johnston, I.C.D.; Stitz, J. Generation of Antibodies Selectively Recognizing Epitopes in a Formaldehyde-Fixed Cell-Surface Antigen Using Virus-like Particle Display and Hybridoma Technology. Antibodies 2023, 12, 57. https://doi.org/10.3390/antib12030057
Schatz S, Willnow L, Winkels M, Rosengarten JF, Theek B, Johnston ICD, Stitz J. Generation of Antibodies Selectively Recognizing Epitopes in a Formaldehyde-Fixed Cell-Surface Antigen Using Virus-like Particle Display and Hybridoma Technology. Antibodies. 2023; 12(3):57. https://doi.org/10.3390/antib12030057
Chicago/Turabian StyleSchatz, Stefanie, Lena Willnow, Monika Winkels, Jamila Franca Rosengarten, Benjamin Theek, Ian C. D. Johnston, and Jörn Stitz. 2023. "Generation of Antibodies Selectively Recognizing Epitopes in a Formaldehyde-Fixed Cell-Surface Antigen Using Virus-like Particle Display and Hybridoma Technology" Antibodies 12, no. 3: 57. https://doi.org/10.3390/antib12030057
APA StyleSchatz, S., Willnow, L., Winkels, M., Rosengarten, J. F., Theek, B., Johnston, I. C. D., & Stitz, J. (2023). Generation of Antibodies Selectively Recognizing Epitopes in a Formaldehyde-Fixed Cell-Surface Antigen Using Virus-like Particle Display and Hybridoma Technology. Antibodies, 12(3), 57. https://doi.org/10.3390/antib12030057