
Functional Activity of Cytokine-Induced Killer Cells Enhanced by CAR-CD19 Modification or by Soluble Bispecific Antibody Blinatumomab

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Primary Cells
2.2. Transposon Plasmids
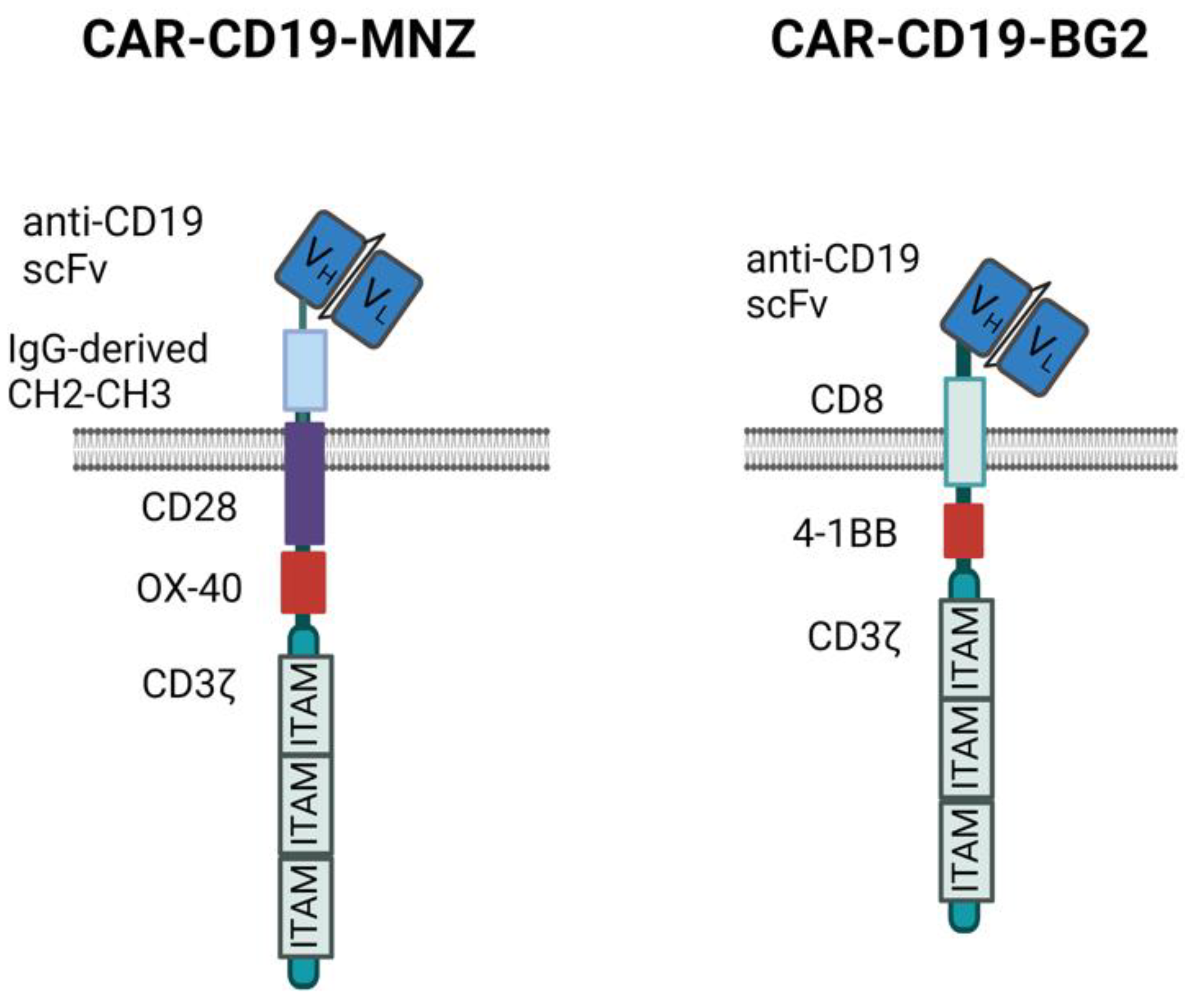
2.3. Generation of Unmodified Cytokine-Induced Killer (CIK) and CARCIK-CD19 Cells
2.4. Flow Cytometry
2.5. Cytotoxicity
2.6. Proliferation
2.7. Intracellular Cytokines
2.8. NF-kB and NFAT Signaling
2.9. Imaging of Immunological Synapse
2.10. In Vivo Animal Model
2.11. Statistical Analyses
3. Results
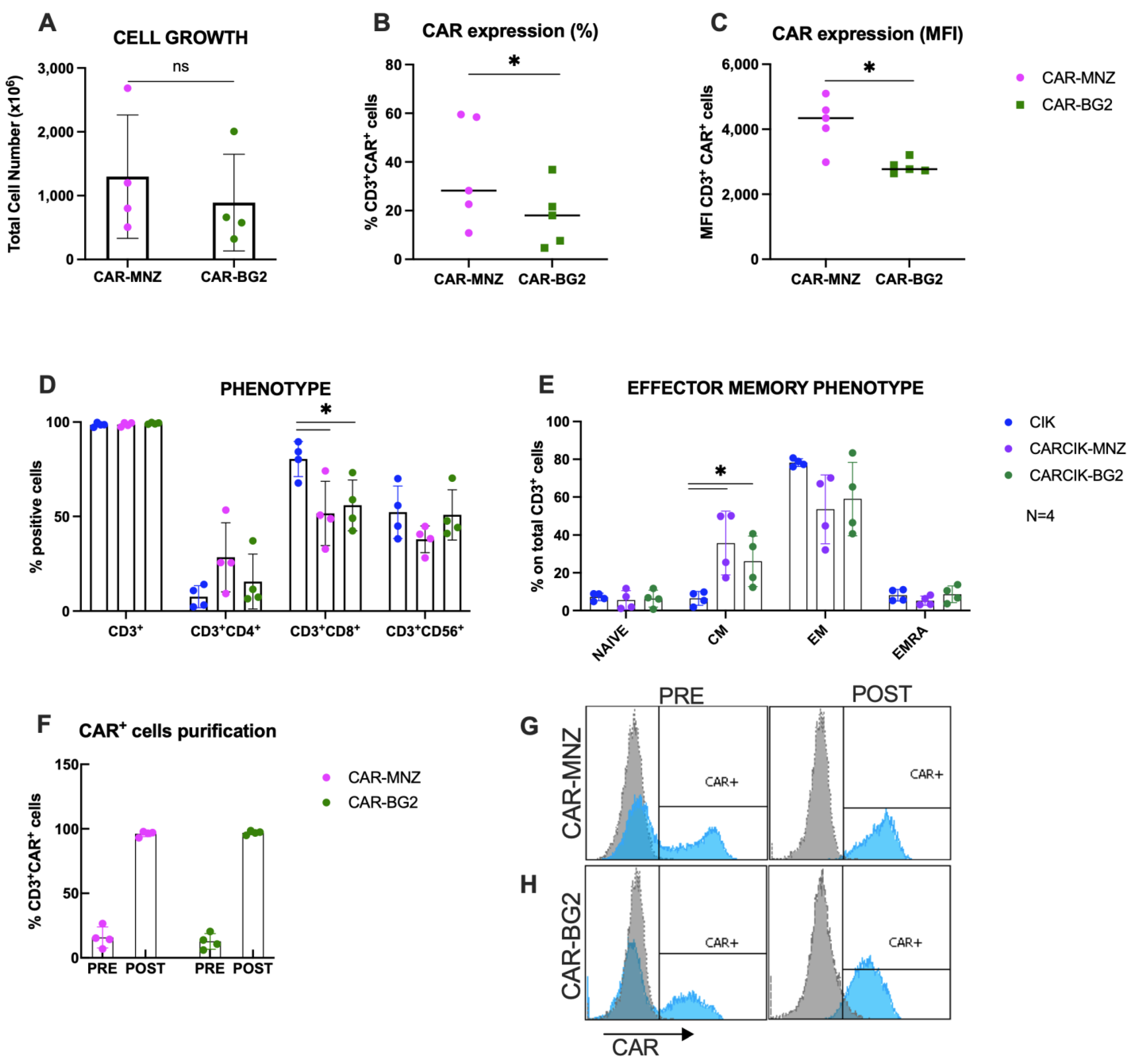
3.1. Characterization of Cytokine-Induced Killer (CIK) and CARCIK-CD19 Cells
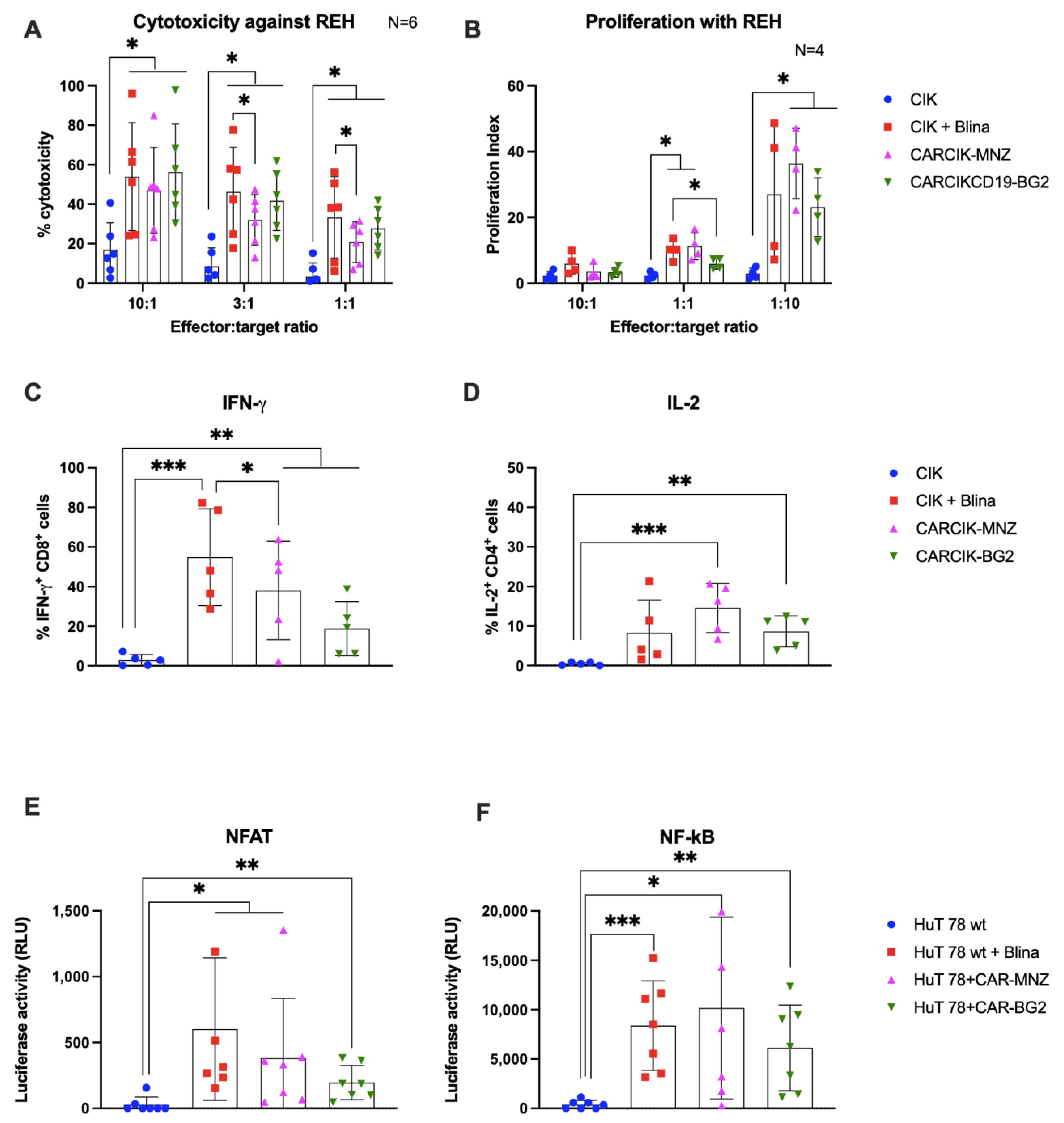
3.2. Cytokine-Induced Killer (CIK) In Vitro Functional Activity Is Enhanced in Presence of Blinatumomab or Anti-CD19 Chimeric Antigen Receptors (CARs)
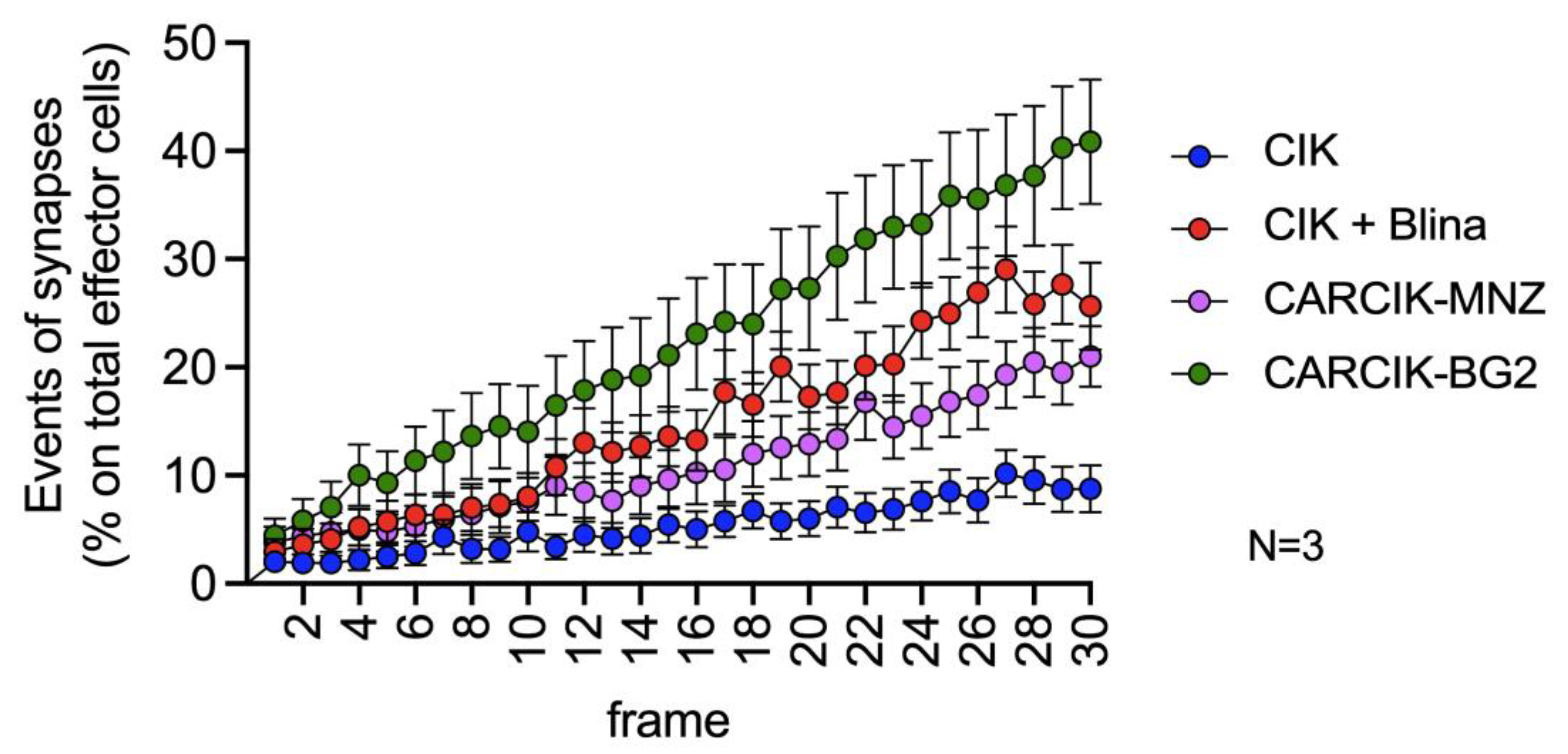
3.3. Chimeric Antigen Receptors (CARs) and Blina Enhance the Immunological Synapse between Cytokine-Induced Killer (CIK) and Target Cells
3.4. Blina and Chimeric Antigen Receptors (CARs) Improve Cytokine-Induced Killer Cells (CIKs) In Vivo Anti-Tumor Efficacy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pievani, A.; Borleri, G.; Pende, D.; Moretta, L.; Rambaldi, A.; Golay, J.; Introna, M. Dual-Functional Capability of CD3 + CD56 + CIK Cells, a T-Cell Subset That Acquires NK Function and Retains TCR-Mediated Specific Cytotoxicity. Blood 2011, 118, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Pievani, A.; Belussi, C.; Klein, C.; Rambaldi, A.; Golay, J.; Introna, M. Enhanced Killing of Human B-Cell Lymphoma Targets by Combined Use of Cytokine-Induced Killer Cell (CIK) Cultures and Anti-CD20 Antibodies. Blood 2011, 117, 510–518. [Google Scholar] [CrossRef]
- Franceschetti, M.; Pievani, A.; Borleri, G.; Vago, L.; Fleischhauer, K.; Golay, J.; Introna, M. Cytokine-Induced Killer Cells Are Terminallydifferentiated Activated CD8 Cytotoxic T-EMRA Lymphocytes. Exp. Hematol. 2009, 37, 616–628.e2. [Google Scholar] [CrossRef]
- Introna, M. CIK as Therapeutic Agents against Tumors. J. Autoimmun. 2017, 85, 32–44. [Google Scholar] [CrossRef]
- Valgardsdottir, R.; Capitanio, C.; Texido, G.; Pende, D.; Cantoni, C.; Pesenti, E.; Rambaldi, A.; Golay, J.; Introna, M. Direct Involvement of CD56 in Cytokine-Induced Killer-Mediated Lysis of CD56+ Hematopoietic Target Cells. Exp. Hematol. 2014, 42, 1013–1021.e1. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Borleri, G.; Conti, E.; Franceschetti, M.; Barbui, A.M.; Broady, R.; Dander, E.; Gaipa, G.; D’Amico, G.; Biagi, E.; et al. Repeated Infusions of Donor-Derived Cytokine-Induced Killer Cells in Patients Relapsing after Allogeneic Stem Cell Transplantation: A Phase I Study. Haematologica 2007, 92, 952–959. [Google Scholar] [CrossRef]
- Lussana, F.; Introna, M.; Golay, J.; Delaini, F.; Pavoni, C.; Valgarsddottir, R.; Gotti, E.; Algarotti, A.; Micò, C.; Grassi, A.; et al. Final Analysis of a Multicenter Pilot Phase 2 Study of Cytokine Induced Killer (CIK) Cells for Patients with Relapse after Allogeneic Transplantation. Blood 2016, 128, 1160. [Google Scholar] [CrossRef]
- Introna, M.; Lussana, F.; Algarotti, A.; Gotti, E.; Valgardsdottir, R.; Micò, C.; Grassi, A.; Pavoni, C.; Ferrari, M.L.; Delaini, F.; et al. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 2070–2078. [Google Scholar] [CrossRef]
- Merker, M.; Salzmann-Manrique, E.; Katzki, V.; Huenecke, S.; Bremm, M.; Bakhtiar, S.; Willasch, A.; Jarisch, A.; Soerensen, J.; Schulz, A.; et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biol. Blood Marrow Transplant. 2019, 25, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Schmeel, L.C.; Schmeel, F.C.; Coch, C.; Schmidt-Wolf, I.G.H. Cytokine-Induced Killer (CIK) Cells in Cancer Immunotherapy: Report of the International Registry on CIK Cells (IRCC). J. Cancer Res. Clin. Oncol. 2015, 141, 839–849. [Google Scholar] [CrossRef]
- Thakur, A.; Sorenson, C.; Norkina, O.; Schalk, D.; Ratanatharathorn, V.; Lum, L.G. Activated T Cells from Umbilical Cord Blood Armed with Anti-CD3 × Anti-CD20 Bispecific Antibody Mediate Specific Cytotoxicity against CD20+ Targets with Minimal Allogeneic Reactivity: A Strategy for Providing Antitumor Effects after Cord Blood Transplants. Transfusion 2012, 52, 63–75. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Vigolo, E.; D’Accardio, G.; Astori, G.; Rosato, A.; Sommaggio, R. How Can Cytokine-Induced Killer Cells Overcome CAR-T Cell Limits. Front. Immunol. 2023, 14, 1229540. [Google Scholar] [CrossRef] [PubMed]
- Tita-Nwa, F.; Moldenhauer, G.; Herbst, M.; Kleist, C.; Ho, A.D.; Kornacker, M. Cytokine-Induced Killer Cells Targeted by the Novel Bispecific Antibody CD19 × CD5 (HD37 × T5.16) Efficiently Lyse B-Lymphoma Cells. Cancer Immunol. Immunother. 2007, 56, 1911–1920. [Google Scholar] [CrossRef]
- Golay, J.; Martinelli, S.; Alzani, R.; Cribioli, S.; Albanese, C.; Gotti, E.; Pasini, B.; Mazzanti, B.; Saccardi, R.; Rambaldi, A.; et al. Cord Blood–Derived Cytokine-Induced Killer Cells Combined with Blinatumomab as a Therapeutic Strategy for CD19 + Tumors. Cytotherapy 2018, 20, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Wang, Y.; Lv, H.; Guo, Y.; Dai, H.; Wicha, M.S.; Chang, A.E.; Li, Q. Cytokine-Induced Killer (CIK) Cells Bound with Anti-CD3/Anti-CD133 Bispecific Antibodies Target CD133(High) Cancer Stem Cells in Vitro and in Vivo. Clin. Immunol. 2013, 149, 156–168. [Google Scholar] [CrossRef]
- Chan, J.K.; Hamilton, C.A.; Cheung, M.K.; Karimi, M.; Baker, J.; Gall, J.M.; Schulz, S.; Thorne, S.H.; Teng, N.N.; Contag, C.H.; et al. Enhanced Killing of Primary Ovarian Cancer by Retargeting Autologous Cytokine-Induced Killer Cells with Bispecific Antibodies: A Preclinical Study. Clin. Cancer Res. 2006, 12, 1859–1867. [Google Scholar] [CrossRef]
- Verneris, M.R.; Arshi, A.; Edinger, M.; Kornacker, M.; Natkunam, Y.; Karami, M.; Cao, Y.; Marina, N.; Contag, C.H.; Negrin, R.S. Low Levels of Her2/Neu Expressed by Ewing’s Family Tumor Cell Lines Can Redirect Cytokine-Induced Killer Cells. Clin. Cancer Res. 2005, 11, 4561–4570. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Nasta, S.; Porter, D.L.; Mato, A.; Shah, G.D.; Landsburg, D.J.; Chong, E.A.; Lacey, S.F.; Melenhorst, J.J.; et al. Phase IIa Trial of Chimeric Antigen Receptor Modified T Cells Directed against CD19 (CTL019) in Patients with Relapsed or Refractory CD19+ Lymphomas. JCO 2015, 33, 8516. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Waldron, E.R.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.; Hamilton, J.; Awasthi, R.; et al. Clinical Pharmacology of Tisagenlecleucel in B-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2018, 24, 6175–6184. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1–2 Trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Ali, A.; Dutta, S.; Banday, S.; Malonia, S.K. Emerging Trends in Immunotherapy for Cancer. Diseases 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Turazzi, N.; Benedicenti, F.; Calabria, A.; Tenderini, E.; Tettamanti, S.; Giordano Attianese, G.M.P.; Cooper, L.J.N.; Aiuti, A.; Montini, E.; et al. Immunotherapy of Acute Leukemia by Chimeric Antigen Receptor-Modified Lymphocytes Using an Improved Sleeping Beauty Transposon Platform. Oncotarget 2016, 7, 51581–51597. [Google Scholar] [CrossRef]
- Magnani, C.F.; Mezzanotte, C.; Cappuzzello, C.; Bardini, M.; Tettamanti, S.; Fazio, G.; Cooper, L.J.N.; Dastoli, G.; Cazzaniga, G.; Biondi, A.; et al. Preclinical Efficacy and Safety of CD19CAR Cytokine-Induced Killer Cells Transfected with Sleeping Beauty Transposon for the Treatment of Acute Lymphoblastic Leukemia. Hum. Gene Ther. 2018, 29, 602–613. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty-Engineered CAR T Cells Achieve Anti-Leukemic Activity without Severe Toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy in Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular Evolution of a Novel Hyperactive Sleeping Beauty Transposase Enables Robust Stable Gene Transfer in Vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef]
- Zaninelli, S.; Meli, C.; Borleri, G.; Quaroni, M.; Pavoni, C.; Gaipa, G.; Biondi, A.; Introna, M.; Golay, J.; Rambaldi, A.; et al. Optimization and Validation of in Vivo Flow Cytometry Chimeric Antigen Receptor T Cell Detection Method Using CD19his Indirect Staining. Cytometry A 2023, 105, 112–123. [Google Scholar] [CrossRef]
- Introna, M.; Franceschetti, M.; Ciocca, A.; Borleri, G.; Conti, E.; Golay, J.; Rambaldi, A. Rapid and Massive Expansion of Cord Blood-Derived Cytokine-Induced Killer Cells: An Innovative Proposal for the Treatment of Leukemia Relapse after Cord Blood Transplantation. Bone Marrow Transplant. 2006, 38, 621–627. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, J.; Kudlacek, S.; Qi, T.; Dunlap, T.; Cao, Y. Population Dynamics of Immunological Synapse Formation Induced by Bispecific T Cell Engagers Predict Clinical Pharmacodynamics and Treatment Resistance. eLife 2023, 12, e83659. [Google Scholar] [CrossRef] [PubMed]
- Kouhestani, D.; Geis, M.; Alsouri, S.; Bumm, T.G.P.; Einsele, H.; Sauer, M.; Stuhler, G. Variant Signaling Topology at the Cancer Cell–T-Cell Interface Induced by a Two-Component T-Cell Engager. Cell Mol. Immunol. 2021, 18, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric Antigen Receptor T Cell Persistence and Memory Cell Formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef]
- Huo, Y.; Sheng, Z.; Lu, D.R.; Ellwanger, D.C.; Li, C.-M.; Homann, O.; Wang, S.; Yin, H.; Ren, R. Blinatumomab-Induced T Cell Activation at Single Cell Transcriptome Resolution. BMC Genom. 2021, 22, 145. [Google Scholar] [CrossRef]
- Stanley, A.C.; Lacy, P. Pathways for Cytokine Secretion. Physiology 2010, 25, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of Regular Cytolytic T Cell Synapses by Bispecific Single-Chain Antibody Constructs on MHC Class I-Negative Tumor Cells. Molecular Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Kent, A.; Longino, N.V.; Christians, A.; Davila, E. Naturally Occurring Genetic Alterations in Proximal TCR Signaling and Implications for Cancer Immunotherapy. Front Immunol. 2021, 12, 658611. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 Costimulation by a Chimeric Antigen Receptor Abrogates CD28 and IL-2 Induced IL-10 Secretion by Redirected CD4 + T Cells. OncoImmunology 2012, 1, 458–466. [Google Scholar] [CrossRef]
- Pulè, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A Chimeric T Cell Antigen Receptor That Augments Cytokine Release and Supports Clonal Expansion of Primary Human T Cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. A Comparison of Chimeric Antigen Receptors Containing CD28 versus 4-1BB Costimulatory Domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Zumerle, S.; Molon, B.; Viola, A. Membrane Rafts in T Cell Activation: A Spotlight on CD28 Costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Liboni, C.; Viola, A. CD28 and Chemokine Receptors: Signalling Amplifiers at the Immunological Synapse. Front. Immunol. 2022, 13, 938004. [Google Scholar] [CrossRef]
- Honikel, M.M.; Olejniczak, S.H. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules 2022, 12, 1303. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric Antigen Receptor T Cells Form Nonclassical and Potent Immune Synapses Driving Rapid Cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-Long Leukaemia Remissions with Persistence of CD4+ CAR T Cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-Independent Activation Enhances the Efficacy of 41BB Co-Stimulated CD22 CAR T Cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Portell, C.A.; Wenzell, C.M.; Advani, A.S. Clinical and Pharmacologic Aspects of Blinatumomab in the Treatment of B-Cell Acute Lymphoblastic Leukemia. Clin. Pharmacol. 2013, 5, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; D’Amico, A.; Borleri, G.; Bonzi, M.; Valgardsdottir, R.; Alzani, R.; Cribioli, S.; Albanese, C.; Pesenti, E.; Finazzi, M.C.; et al. A Novel Method Using Blinatumomab for Efficient, Clinical-Grade Expansion of Polyclonal T Cells for Adoptive Immunotherapy. J. Immunol. 2014, 193, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Ramírez-Fernández, Á.; Bueno, C.; Argemí-Muntadas, L.; Fuentes, P.; Aguilar-Sopeña, Ó.; Gutierrez-Agüera, F.; Zanetti, S.R.; Tapia-Galisteo, A.; Díez-Alonso, L.; et al. Overcoming CAR-Mediated CD19 Downmodulation and Leukemia Relapse with T Lymphocytes Secreting Anti-CD19 T-Cell Engagers. Cancer Immunol. Res. 2022, 10, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape without Detectable Toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Van der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Guercio, M.; Orlando, D.; Di Cecca, S.; Sinibaldi, M.; Boffa, I.; Caruso, S.; Abbaszadeh, Z.; Camera, A.; Cembrola, B.; Bovetti, K.; et al. CD28.OX40 Co-Stimulatory Combination Is Associated with Long in Vivo Persistence and High Activity of CAR.CD30 T Cells. Haematologica 2020, 106, 987–999. [Google Scholar] [CrossRef]
- Quintarelli, C.; Orlando, D.; Boffa, I.; Guercio, M.; Polito, V.A.; Petretto, A.; Lavarello, C.; Sinibaldi, M.; Weber, G.; Del Bufalo, F.; et al. Choice of Costimulatory Domains and of Cytokines Determines CAR T-Cell Activity in Neuroblastoma. Oncoimmunology 2018, 7, e1433518. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-Shelf” Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaninelli, S.; Panna, S.; Tettamanti, S.; Melita, G.; Doni, A.; D’Autilia, F.; Valgardsdottir, R.; Gotti, E.; Rambaldi, A.; Golay, J.; et al. Functional Activity of Cytokine-Induced Killer Cells Enhanced by CAR-CD19 Modification or by Soluble Bispecific Antibody Blinatumomab. Antibodies 2024, 13, 71. https://doi.org/10.3390/antib13030071
Zaninelli S, Panna S, Tettamanti S, Melita G, Doni A, D’Autilia F, Valgardsdottir R, Gotti E, Rambaldi A, Golay J, et al. Functional Activity of Cytokine-Induced Killer Cells Enhanced by CAR-CD19 Modification or by Soluble Bispecific Antibody Blinatumomab. Antibodies. 2024; 13(3):71. https://doi.org/10.3390/antib13030071
Chicago/Turabian StyleZaninelli, Silvia, Silvia Panna, Sarah Tettamanti, Giusi Melita, Andrea Doni, Francesca D’Autilia, Rut Valgardsdottir, Elisa Gotti, Alessandro Rambaldi, Josée Golay, and et al. 2024. "Functional Activity of Cytokine-Induced Killer Cells Enhanced by CAR-CD19 Modification or by Soluble Bispecific Antibody Blinatumomab" Antibodies 13, no. 3: 71. https://doi.org/10.3390/antib13030071
APA StyleZaninelli, S., Panna, S., Tettamanti, S., Melita, G., Doni, A., D’Autilia, F., Valgardsdottir, R., Gotti, E., Rambaldi, A., Golay, J., & Introna, M. (2024). Functional Activity of Cytokine-Induced Killer Cells Enhanced by CAR-CD19 Modification or by Soluble Bispecific Antibody Blinatumomab. Antibodies, 13(3), 71. https://doi.org/10.3390/antib13030071