
Purification of a Fc-Fusion Protein with [Bathophenathroline:metal] Complexes

 , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of 0.2M Bathophenanthroline:DMSO:HCl (Batho:DMSO:HCl) Solution
2.2.2. Preparation of AChE-Fc Fusion Proteins
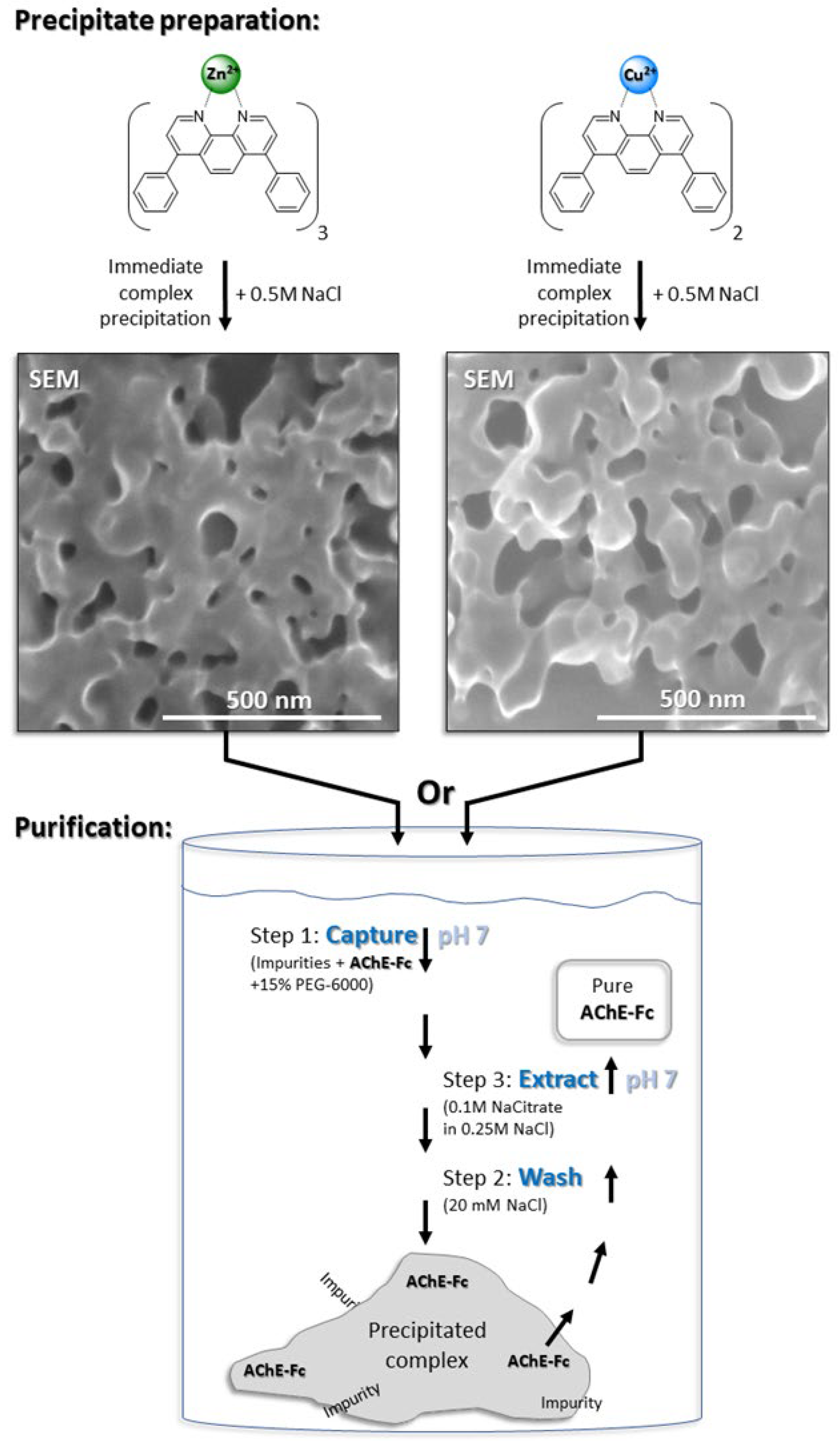
2.2.3. Purification of AChE-Fc via [(batho)3:Zn2+] or [(batho)2:Cu2+] Complexes
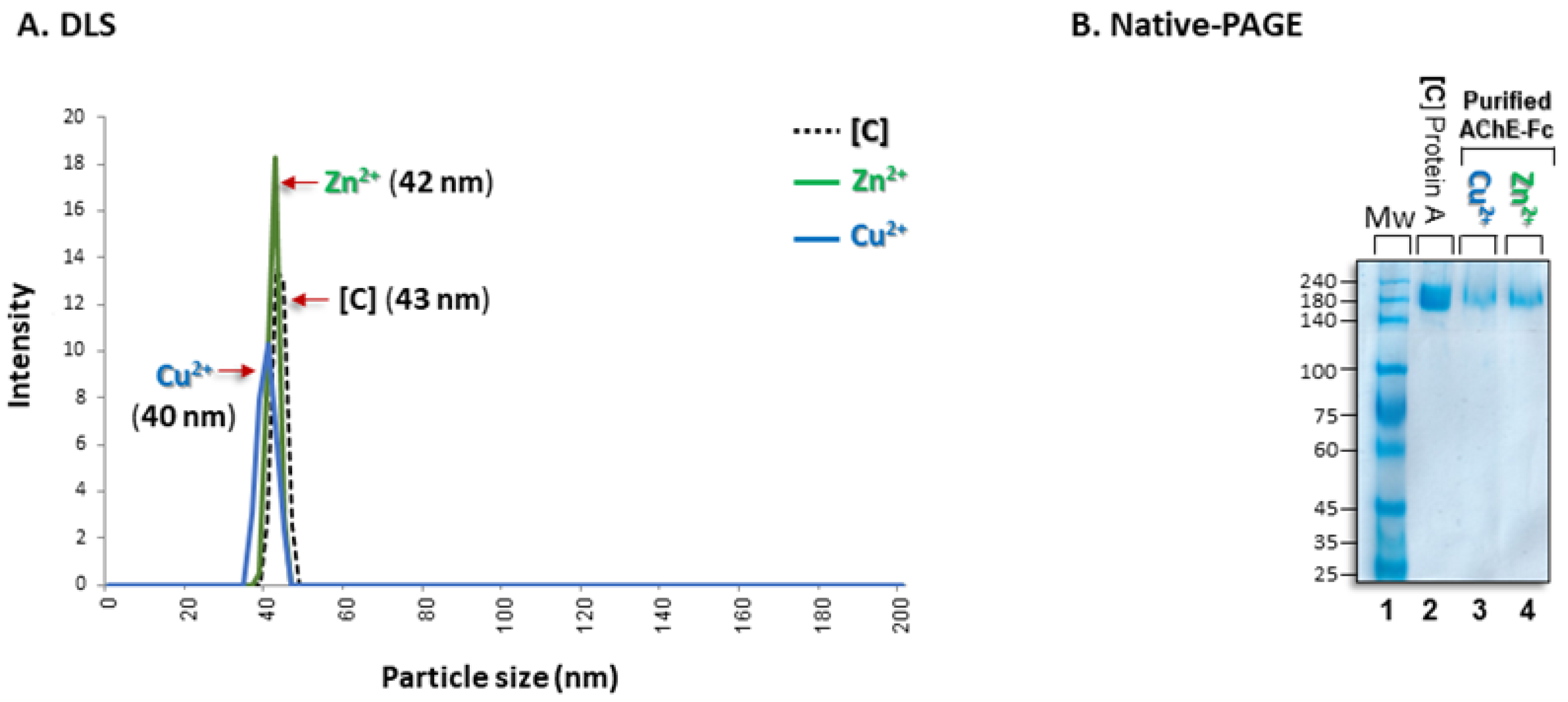
2.2.4. Dynamic Light Scattering (DLS)
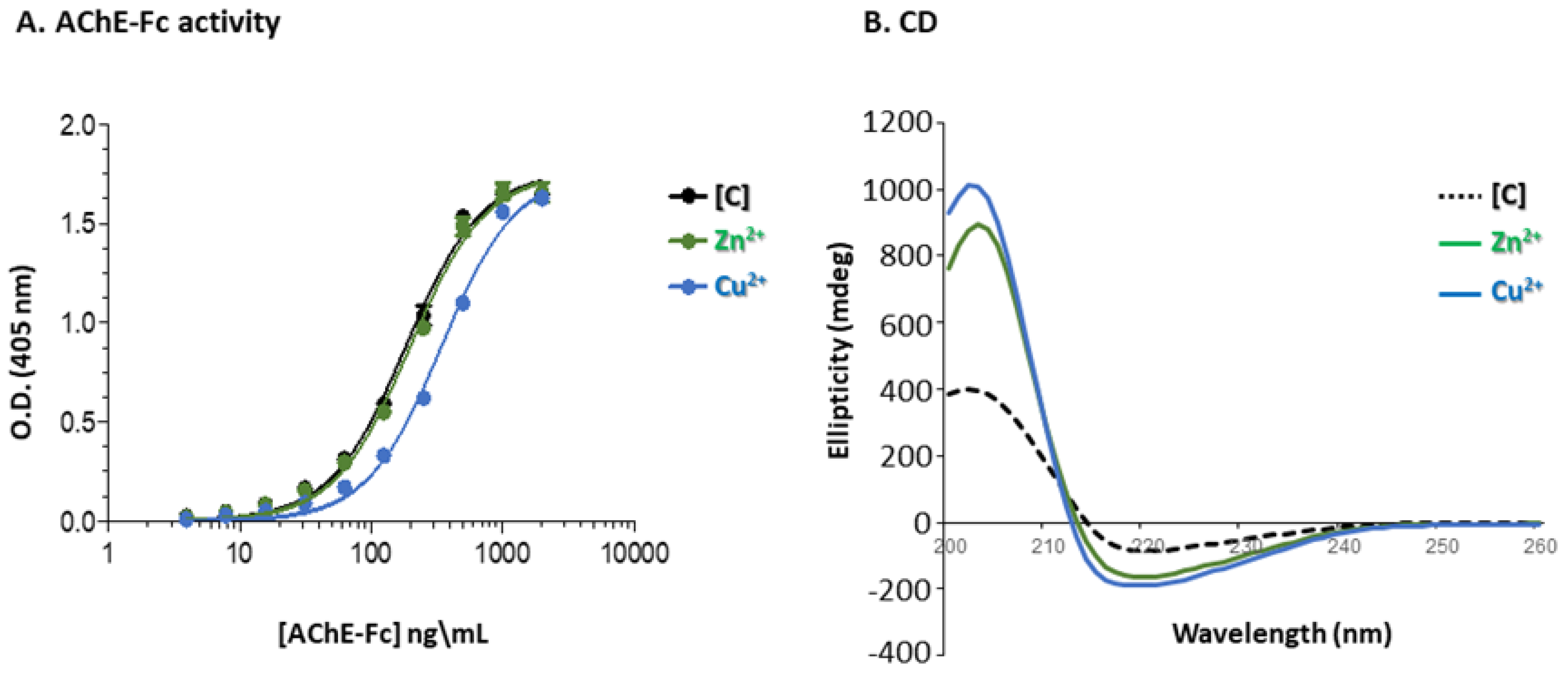
2.2.5. Circular Dichroism (CD) Spectroscopy
2.2.6. Native PAGE
2.2.7. Batho Recrystallization and Quantitation
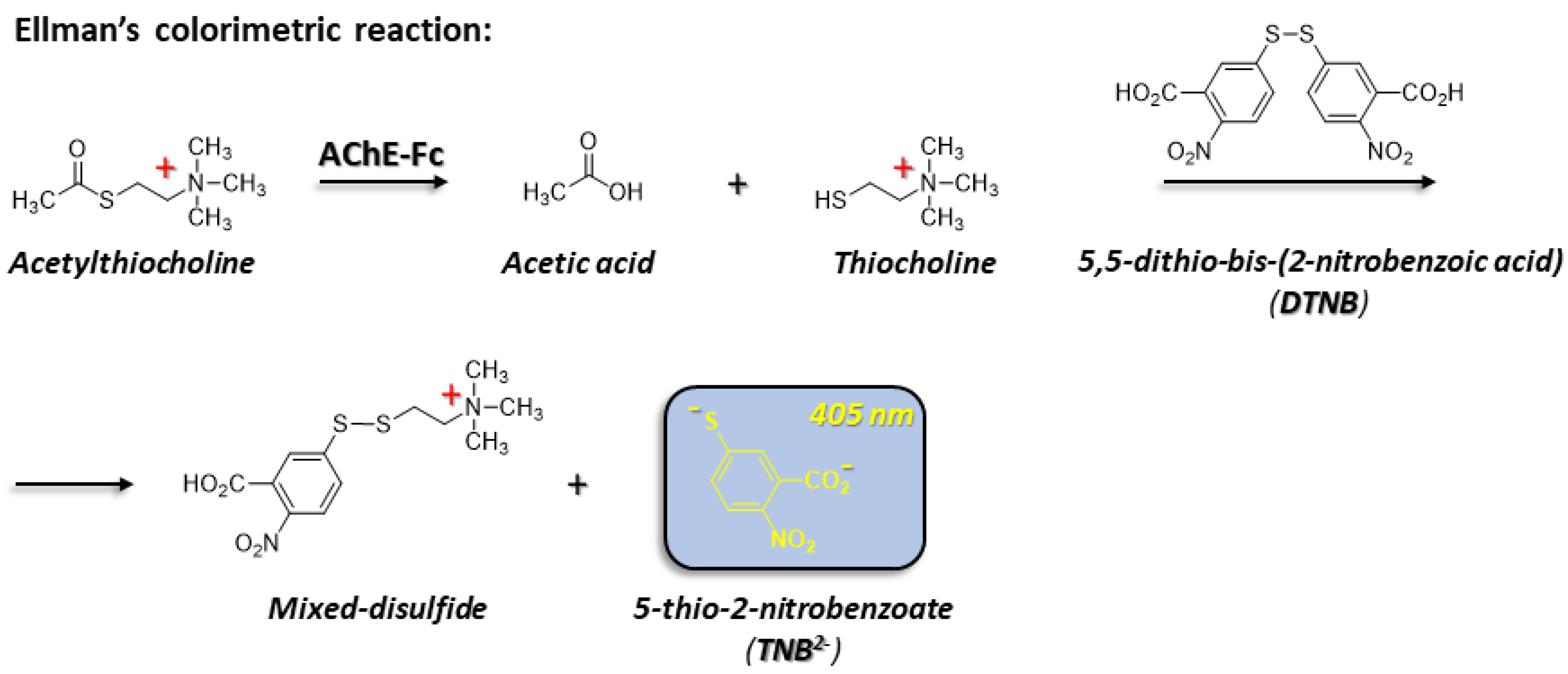
2.2.8. AChE-Fc Activity
2.2.9. Leaching Assessment
2.2.10. Scanning Electron Microscopy (SEM)
3. Results and Discussion
3.1. Precipitation of the Metal-Chelator Complexes
3.2. The Role of PEG 6000
3.3. Aggregational State of the Recovered Fc-AChE
3.4. Enzymatic Activity
3.5. Binding Interactions and Process Yield
3.6. Binding Capacity
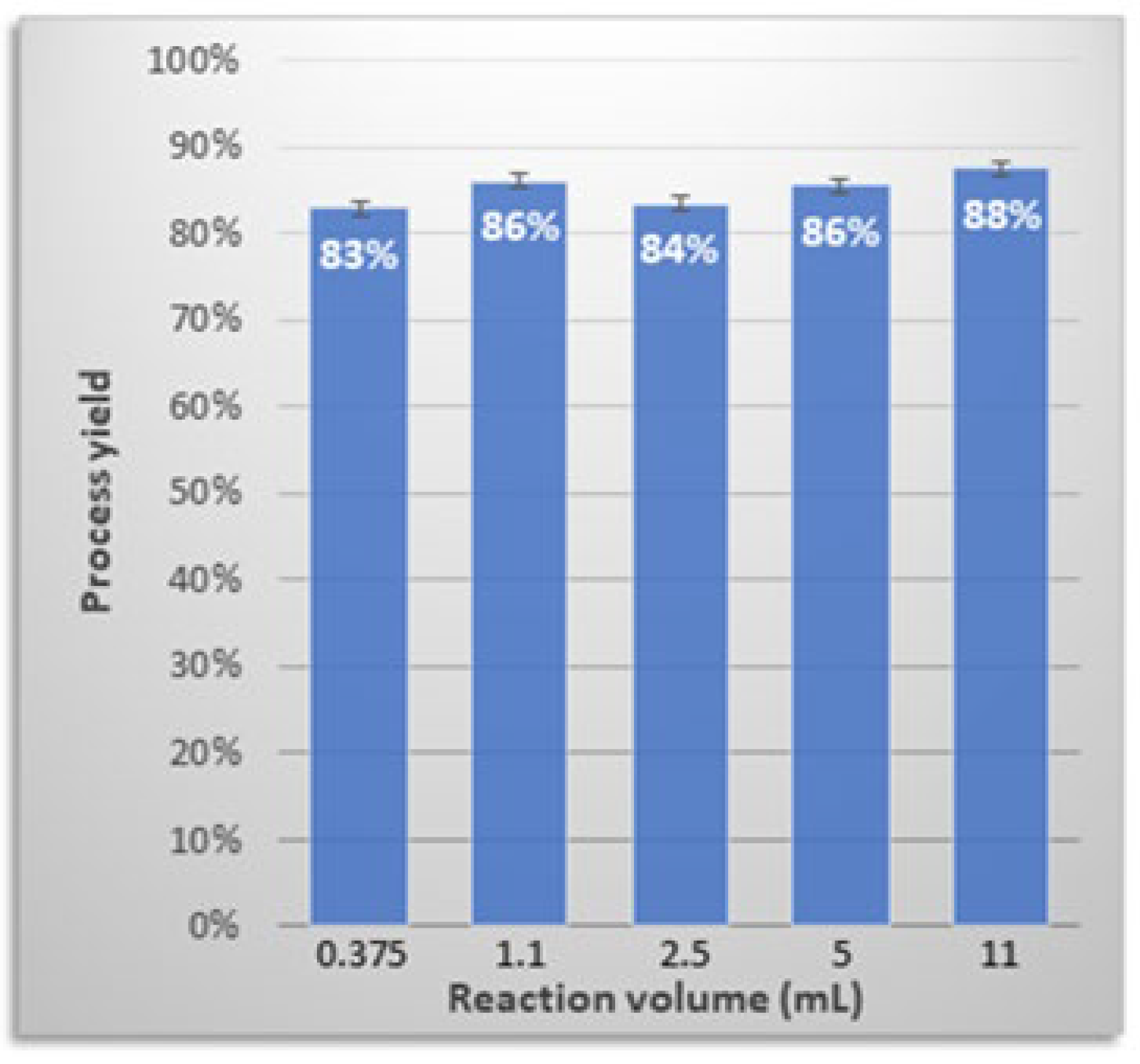
3.7. Process Upscaling
3.8. Chelator Recycling
3.9. Estimated Chelator Leaching Under Extraction Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capon, D.J.; Chamow, S.M.; Mordenti, J.; Marsters, S.A.; Gregory, T.; Mitsuya, H.; Byrn, R.A.; Lucas, C.; Wurm, F.M.; Groopman, J.E. Designing CD4 immunoadhesins for AIDS therapy. Nature 1989, 337, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Wedzina, A.S.; Borek, A.; Chudzian, J.; Jakimowicz, P.; Zakrzewska, M.; Otlewski, J. Efficient production and purification of extracellular domain of human FGFR-Fc fusion proteins from Chinese hamster ovary cells. Protein Expr. Purif. 2014, 99, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; He, B.; Zhou, P.; Derda, R.; Huang, J. Molecular Design of Peptide-Fc Fusion Drugs. Curr. Drug Metab. 2019, 20, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.A.; Huang, A.J.; Pan, C.Q.; Lazarus, R.A. Expression and Characterization of a DNase I-Fc Fusion Enzyme*. J. Biol. Chem. 1999, 274, 9738–9743. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Cohen, O.; Ehrlich, S.; Epstein, E.; Alcalay, R.; Mazor, O. Acetylcholinesterase-Fc Fusion Protein (AChE-Fc): A Novel Potential Organophosphate Bioscavenger with Extended Plasma Half-Life. Bioconjug. Chem. 2015, 26, 1753–1758. [Google Scholar] [CrossRef]
- Jazayeri, J.A.; Carroll, G.J. Fc-Based Cytokines. BioDrugs 2008, 22, 11–26. [Google Scholar] [CrossRef]
- Duivelshof, B.L.; Murisier, A.; Camperi, J.; Fekete, S.; Beck, A.; Guillarme, D.; D‘Atri, V. Therapeutic Fc-fusion proteins: Current analytical strategies. J. Sep. Sci. 2021, 44, 35–62. [Google Scholar] [CrossRef]
- Zhang, J.; Carter, J.; Siu, S.; O‘Neill, J.W.; Gates, H.A.; Delaney, J.; Mehlin, C. Fusion partners as a tool for the expression of difficult proteins in mammalian cells. Curr. Pharm. Biotechnol. 2010, 11, 241–245. [Google Scholar] [CrossRef]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef]
- Meibohm, B.; Zhou, H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J. Clin. Pharmacol. 2012, 52 (Suppl. S1), 54s–62s. [Google Scholar] [CrossRef]
- Nardella, F.A.; Teller, D.C. Fc intermediate (Fci), a papain-generated fragment of human IgG, intermediate in charge, molecular weight and cleavage between the Fc and Fc’ fragments of IgG. Mol. Immunol. 1985, 22, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Jevsevar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.; Park, S.; Sohn, M.H.; Jo, M.; Ko, B.J.; Na, J.H.; Yoo, H.; Jeong, A.L.; Ha, K.; Woo, J.R. An Fc variant with two mutations confers prolonged serum half-life and enhanced effector functions on IgG antibodies. Exp. Mol. Med. 2022, 54, 1850–1861. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Rath, T.; Baker, K.; Dumont, J.A.; Peters, R.T.; Jiang, H.; Qiao, S.W.; Lencer, W.I.; Pierce, G.F.; Blumberg, R.S. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015, 35, 235–254. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Weflen, A.W.; Baier, N.; Tang, Q.J.; Hof, M.V.D.; Blumberg, R.S.; Lencer, W.I.; Massol, R.H. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol. Biol. Cell 2013, 24, 2398–2405. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Mekhaiel, D.N.; Czajkowsky, D.M.; Andersen, J.T.; Shi, J.; El-Faham, M.; Doenhoff, M.; McIntosh, R.S.; Sandlie, I.; He, J.; Hu, J. Polymeric human Fc-fusion proteins with modified effector functions. Sci. Rep. 2011, 1, 124. [Google Scholar] [CrossRef]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Kanje, S. Chapter 2—Engineering of Protein A for improved purification of antibodies and Fc-fused proteins. In Approaches to the Purification, Analysis and Characterization of Antibody-Based Therapeutics; Matte, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 35–54. [Google Scholar]
- Roque, A.C.; Silva, C.S.; Taipa, M.A. Affinity-based methodologies and ligands for antibody purification: Advances and perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar] [CrossRef]
- Tscheliessnig, A.; Satzer, P.; Hammerschmidt, N.; Schulz, H.; Helk, B.; Jungbauer, A. Ethanol precipitation for purification of recombinant antibodies. J. Biotechnol. 2014, 188, 17–28. [Google Scholar] [CrossRef]
- Mao, L.N.; Rogers, J.K.; Westoby, M.; Conley, L.; Pieracci, J. Downstream antibody purification using aqueous two-phase extraction. Biotechnol. Prog. 2010, 26, 1662–1670. [Google Scholar] [CrossRef]
- Smejkal, B.; Agrawal, N.J.; Helk, B.; Schulz, H.; Giffard, M.; Mechelke, M.; Ortner, F.; Heckmeier, P.; Trout, B.L.; Hekmat, D. Fast and scalable purification of a therapeutic full-length antibody based on process crystallization. Biotechnol. Bioeng. 2013, 110, 2452–2461. [Google Scholar] [CrossRef]
- Bolton, G.R.; Mehta, K.K. The role of more than 40 years of improvement in protein A chromatography in the growth of the therapeutic antibody industry. Biotechnol. Prog. 2016, 32, 1193–1202. [Google Scholar] [CrossRef]
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 40–47. [Google Scholar] [CrossRef]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, M.; Montelione, G.T. Structures of bacterial immunoglobulin-binding domains and their complexes with immunoglobulins. Curr. Opin. Struct. Biol. 1995, 5, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, M.; Tejero, R.; Zimmerman, D.E.; Celda, B.; Nilsson, B.; Montelione, G.T. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol. 1997, 272, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef]
- Hale, G.; Drumm, A.; Harrison, P.; Phillips, J. Repeated cleaning of protein A affinity column with sodium hydroxide. J. Immunol. Methods 1994, 171, 15–21. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hinckley, P. Host cell protein clearance during protein A chromatography: Development of an improved column wash step. Biotechnol. Prog. 2008, 24, 1115–1121. [Google Scholar] [CrossRef]
- Pabst, T.M.; Thai, J.; Hunter, A.K. Evaluation of recent Protein A stationary phase innovations for capture of biotherapeutics. J. Chromatogr. A 2018, 1554, 45–60. [Google Scholar] [CrossRef]
- Liu, Z.; Mostafa, S.S.; Shukla, A.A. A comparison of protein A chromatographic stationary phases: Performance characteristics for monoclonal antibody purification. Biotechnol. Appl. Biochem. 2015, 62, 37–47. [Google Scholar] [CrossRef]
- Hahn, R.; Schlegel, R.; Jungbauer, A. Comparison of protein A affinity sorbents. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 790, 35–51. [Google Scholar] [CrossRef]
- Pfaunmiller, E.L.; Paulemond, M.L.; Dupper, C.M.; Hage, D.S. Affinity monolith chromatography: A review of principles and recent analytical applications. Anal. Bioanal. Chem. 2013, 405, 2133–2145. [Google Scholar] [CrossRef]
- Boi, C. Membrane adsorbers as purification tools for monoclonal antibody purification. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S. Process economics of industrial monoclonal antibody manufacture. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Love, J.C.; Love, K.R.; Barone, P.W. Enabling global access to high-quality biopharmaceuticals. Curr. Opin. Chem. Eng. 2013, 2, 383–390. [Google Scholar] [CrossRef]
- Withanage, T.J.; Lal, M.; Salem, H.; Krichevski, O.; Wachtel, E.; Patchornik, G. The [(bathophenanthroline)(3):Fe(2+)] complex as an aromatic non-polymeric medium for purification of human lactoferrin. J. Chromatogr. A 2024, 1732, 465218. [Google Scholar] [CrossRef]
- Trudel, J.; Asselin, A. Protein purification for microsequencing by sequential native and denaturing polyacrylamide gel electrophoresis: Application to one chitinase. Anal. Biochem. 1994, 221, 214–216. [Google Scholar] [CrossRef]
- Smith, G.L.; Reutovich, A.A.; Srivastava, A.K.; Reichard, R.E.; Welsh, C.H.; Melman, A.; Bou-Abdallah, F. Complexation of ferrous ions by ferrozine, 2,2′-bipyridine and 1,10-phenanthroline: Implication for the quantification of iron in biological systems. J. Inorg. Biochem. 2021, 220, 111460. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andreas, V., Jr.; Feather-stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- O’Laughlin, J.W. Separation of cationic metal chelates of 1,10-phenanthroline by liquid chromatography. Anal. Chem. 1982, 54, 178–181. [Google Scholar] [CrossRef]
- Ng, N.S.; Wu, M.J.; Jones, C.E.; Aldrich-Wright, J.R. The antimicrobial efficacy and DNA binding activity of the copper(II) complexes of 3,4,7,8-tetramethyl-1,10-phenanthroline, 4,7-diphenyl-1,10-phenanthroline and 1,2-diaminocyclohexane. J. Inorg. Biochem. 2016, 162, 62–72. [Google Scholar] [CrossRef]
- Hellawell, J.M. Biological Indicators of Freshwater Pollution and Environmental Management; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Dhandapani, G.; Wachtel, E.; Sheves, M.; Patchornik, G. Nonionic detergent micelle aggregates: An economical alternative to protein A chromatography. New Biotechnol. 2021, 61, 90–98. [Google Scholar] [CrossRef]
- Lavery, P.E.; Kowalczykowski, S.C. Enhancement of recA protein-promoted DNA strand exchange activity by volume-occupying agents. J. Biol. Chem. 1992, 267, 9307–9314. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bowman, B.; Tu, S.; Nykypanchuk, D.; Kuksenok, O.; Minko, S. Polyethylene Glycol Crowder’s Effect on Enzyme Aggregation, Thermal Stability, and Residual Catalytic Activity. Langmuir 2021, 37, 8474–8485. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Guo, Z. Effects of Macromolecular Crowding on the Intrinsic Catalytic Efficiency and Structure of Enterobactin-Specific Isochorismate Synthase. J. Am. Chem. Soc. 2007, 129, 730–731. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Takahashi, K.; Kaizu, K.; Matsuda, M. A quantitative model of ERK MAP kinase phosphorylation in crowded media. Sci. Rep. 2013, 3, 1541. [Google Scholar] [CrossRef]
- Khodabandehloo, A.; Chen, D.D.Y. Particle Sizing Methods for The Detection of Protein Aggregates in Biopharmaceuticals. Bioanalysis 2017, 9, 313–326. [Google Scholar] [CrossRef]
- Nobbmann, U.; Connah, M.; Fish, B.; Varley, P.; Gee, C.; Mulot, S.; Chen, J.; Zhou, L.; Lu, Y.; Shen, F. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 2007, 24, 117–128. [Google Scholar] [CrossRef]
- Eyer, P.; Worek, F.; Kiderlen, D.; Sinko, G.; Stuglin, A.; Simeon-Rudolf, V.; Reiner, E. Molar absorption coefficients for the reduced Ellman reagent: Reassessment. Anal. Biochem. 2003, 312, 224–227. [Google Scholar] [CrossRef]
- Smith, R.M.; Martell, A.E. Critical Stability Constants: Volume 2: Amines; Springer: Berlin/Heidelberg, Germany, 1975. [Google Scholar]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Tetin, S.Y.; Prendergast, F.G.; Venyaminov, S.Y. Accuracy of protein secondary structure determination from circular dichroism spectra based on immunoglobulin examples. Anal. Biochem. 2003, 321, 183–187. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Bruque, M.G.; Rodger, A.; Hoffmann, S.V.; Jones, N.C.; Aucamp, J.; Dafforn, T.R.; Thomas, O.R.T. Analysis of the Structure of 14 Therapeutic Antibodies Using Circular Dichroism Spectroscopy. Anal. Chem. 2024, 96, 15151–15159. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Seidl, A.; Sörgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, G.; Howard, A.; Truong, T.V.; Baiju, T.V.; Kesselman, E.; Friedman, N.; Wachtel, E.; Sheves, M.; Danino, D.; Namboothiri, I.N.N. A general platform for antibody purification utilizing engineered-micelles. MAbs 2019, 11, 583–592. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Withanage, T.J.; Alcalay, R.; Krichevsky, O.; Wachtel, E.; Mazor, O.; Patchornik, G. Purification of a Fc-Fusion Protein with [Bathophenathroline:metal] Complexes. Antibodies 2025, 14, 11. https://doi.org/10.3390/antib14010011
Withanage TJ, Alcalay R, Krichevsky O, Wachtel E, Mazor O, Patchornik G. Purification of a Fc-Fusion Protein with [Bathophenathroline:metal] Complexes. Antibodies. 2025; 14(1):11. https://doi.org/10.3390/antib14010011
Chicago/Turabian StyleWithanage, Thisara Jayawickrama, Ron Alcalay, Olga Krichevsky, Ellen Wachtel, Ohad Mazor, and Guy Patchornik. 2025. "Purification of a Fc-Fusion Protein with [Bathophenathroline:metal] Complexes" Antibodies 14, no. 1: 11. https://doi.org/10.3390/antib14010011
APA StyleWithanage, T. J., Alcalay, R., Krichevsky, O., Wachtel, E., Mazor, O., & Patchornik, G. (2025). Purification of a Fc-Fusion Protein with [Bathophenathroline:metal] Complexes. Antibodies, 14(1), 11. https://doi.org/10.3390/antib14010011