
Challenges and Insights in Absolute Quantification of Recombinant Therapeutic Antibodies by Mass Spectrometry: An Introductory Review



Abstract
:1. Historical Overview: Evolution of Recombinant mAbs

2. Current Strategies in MS-Based Quantification of Antibodies
2.1. Quantification of Enzymatically Digested Antibodies
2.1.1. Purification and Enrichment
2.1.2. Denaturation, Reduction, and Alkylation
2.1.3. Digestion
2.1.4. Signature Peptide Selection
2.2. Quantification of Intact Antibodies
2.3. Quantification of Hydrolysed Antibodies
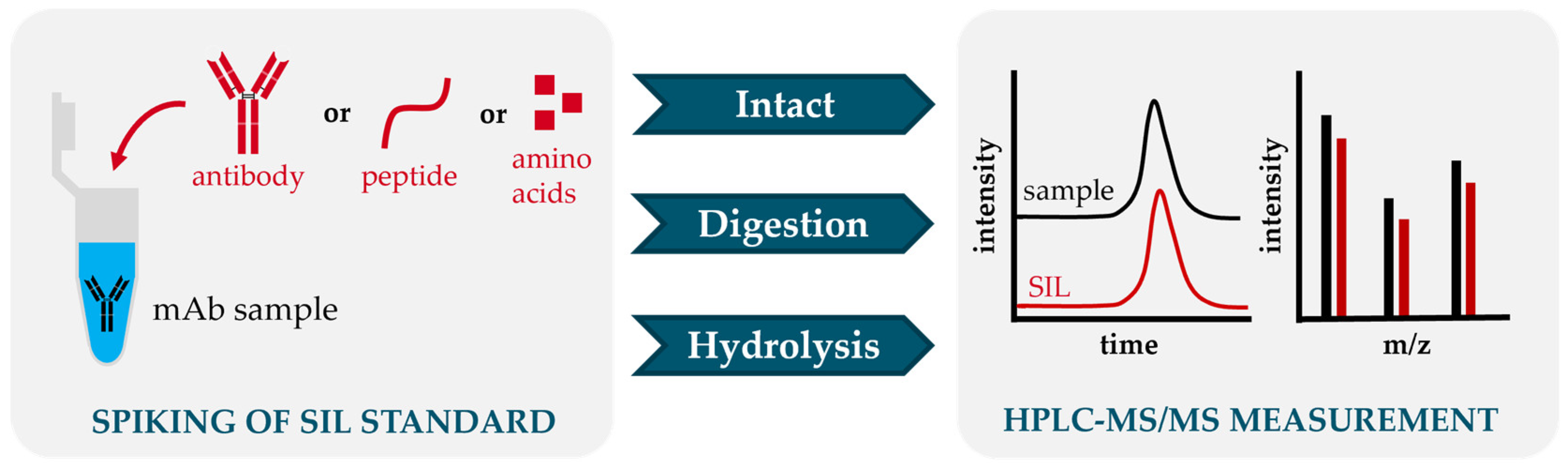
3. Selection of Internal Standards for Quantification with HPLC-MS/MS
3.1. Intact Antibody Standards
3.2. Peptide Standards
3.3. Amino Acid Standards
4. Software Tools Supporting Targeted mAb Quantification
4.1. Commercial and Device-Specific Software
4.2. Open-Source Software Alternatives
5. Outlook: Need for Standardized Protocols, Certified Reference Materials, and New Technologies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Acrylamide |
| AAA | Amino acid analysis |
| ABCD | AntiBodies Chemically Defined |
| ADC | Antibody-drug conjugate |
| AI | Artificial intelligence |
| AIST | National Institute of Advanced Industrial Science and Technology |
| BLAST | Basic Local Alignment Search Tool |
| CAM | Chloroacetamide |
| CDR | Complementarity-determining region |
| CH1-3 | Constant heavy chains 1 to 3 |
| CHO | Chinese hamster ovary |
| CL | Constant light chain |
| CRM | Certified reference material |
| CV | Coefficient of variation |
| DAR | Drug-antibody ratio |
| DOC | Sodium dodecyl sulfate |
| DTT | Dithiothreitol |
| EMA | European Medicines Agency |
| ESI | Electrospray ionization |
| FAB | Antigen-binding sites of antibody |
| FC | Fragment crystallizable region |
| FDA | Food and Drug Administration |
| HAMA | Human anti-mouse antibody |
| HILIC | Hydrophilic interaction chromatography |
| HPLC-MS/MS | High-performance liquid chromatography-tandem mass spectrometry |
| HRMS | High-resolution mass spectrometry |
| IAA | Iodoacetic acid |
| IAC | Immunoaffinity capture |
| IAM | Iodoacetamide |
| ID-MS | Isotope dilution mass spectrometry |
| IEX | Ion exchange |
| IgG1 | Immunoglobulin G antibody class 1 |
| IgG4 | Immunoglobulin G antibody class 4 |
| IL-12 | Interleukin-12 |
| IL-23 | Interleukin-23 |
| IMGT | ImmunoGenetics Information system |
| K | Lysine |
| LBA | Ligand binding assay |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| mAb | Therapeutic monoclonal antibody |
| MAM | Multi-attribute method |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry |
| NIST | National Institute of Standards and Technology |
| NMI | National Metrology Institute |
| opt. | Optional |
| PD-1 | Programmed cell death protein 1 |
| PSAQ | Protein standard absolute quantification |
| PTM | Post-translational modification |
| PURE | Protein synthesis Using Recombinant Elements |
| QC | Quality control |
| QqQ | Triple-quadrupole |
| Q-Trap | Quadrupole-ion trap |
| R | Arginine |
| RP | Reversed phase |
| RSD | Relative standard deviation |
| SDS | Sodium dodecyl sulfate |
| SI units | International standard system of units |
| SIL | Stable isotope-labeled |
| SPE | Solid-phase extraction |
| TCEP | Tris(2-carboxyethyl)phosphine |
| tMRM | Triggered MRM |
| TNF-α | Tumor necrosis factor-alpha |
| TPCK | Tosyl-phenylalanyl-chloromethyl-ketone |
| VH | Variable heavy chain |
| VL | Variable light chain |
| WHO | World Health Organization |
| XIC | Extracted ion chromatogram |
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.A.; Brogden, R.N. Muromonab CD3. A review of its pharmacology and therapeutic potential. Drugs 1989, 37, 871–899. [Google Scholar] [CrossRef] [PubMed]
- Schroff, R.W.; Foon, K.A.; Beatty, S.M.; Oldham, R.K.; Morgan, A.C., Jr. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res. 1985, 45, 879–885. [Google Scholar] [PubMed]
- Jaffers, G.J.; Fuller, T.; Cosimi, A.; Russell, P.; Winn, H.; Colvin, R. Monoclonal antibody therapy. Anti-idiotypic and non-anti-idiotypic antibodies to OKT3 arising despite intense immunosuppression. Transplantation 1986, 41, 572–578. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef]
- Brüggemann, M.; Winter, G.; Waldmann, H.; Neuberger, M.S. The immunogenicity of chimeric antibodies. J. Exp. Med. 1989, 170, 2153–2157. [Google Scholar] [CrossRef]
- Faulds, D.; Sorkin, E.M. Abciximab (c7E3 Fab). A review of its pharmacology and therapeutic potential in ischaemic heart disease. Drugs 1994, 48, 583–598. [Google Scholar] [CrossRef]
- Leget, G.A.; Czuczman, M.S. Use of rituximab, the new FDA-approved antibody. Curr. Opin. Oncol. 1998, 10, 548–551. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef]
- Wiseman, L.R.; Faulds, D. Daclizumab: A review of its use in the prevention of acute rejection in renal transplant recipients. Drugs 1999, 58, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Zhang, Y. Evolution of phage display libraries for therapeutic antibody discovery. mAbs 2023, 15, 2213793. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef]
- Sánchez-Robles, E.M.; Girón, R.; Paniagua, N.; Rodríguez-Rivera, C.; Pascual, D.; Goicoechea, C. Monoclonal Antibodies for Chronic Pain Treatment: Present and Future. Int. J. Mol. Sci. 2021, 22, 10325. [Google Scholar] [CrossRef]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.-C.; Mashayekh, R.; Wymore, K.; McCabe, J.G.; et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994, 368, 856–859. [Google Scholar] [CrossRef]
- Jakobovits, A.; Amado, R.G.; Yang, X.; Roskos, L.; Schwab, G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat. Biotechnol. 2007, 25, 1134–1143. [Google Scholar] [CrossRef]
- Lapadula, G.; Marchesoni, A.; Armuzzi, A.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimaz, R.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; et al. Adalimumab in the Treatment of Immune-Mediated Diseases. Int. J. Immunopathol. Pharmacol. 2014, 27, 33–48. [Google Scholar] [CrossRef]
- Verdin, P. Top companies and drugs by sales in 2023. Nat. Rev. Drug Discov. 2024, 23, 240. [Google Scholar] [CrossRef] [PubMed]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.-O. Current Status and Future Directions of the Immune Checkpoint Inhibitors Ipilimumab, Pembrolizumab, and Nivolumab in Oncology. Ann. Pharmacother. 2015, 49, 907–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, M.; Li, Y.; Li, Y.; Chen, Z.; Tang, Y.; Yang, M.; Deng, G.; Liu, H. A review of the clinical efficacy of FDA-approved antibody–drug conjugates in human cancers. Mol. Cancer 2024, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef]
- Zhu, X.; Huo, S.; Xue, C.; An, B.; Qu, J. Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. J. Pharm. Anal. 2020, 10, 209–220. [Google Scholar] [CrossRef]
- Todoroki, K.; Yamada, T.; Mizuno, H.; TOYO’OKA, T. Current mass spectrometric tools for the bioanalyses of therapeutic monoclonal antibodies and antibody-drug conjugates. Anal. Sci. 2018, 34, 397–406. [Google Scholar] [CrossRef]
- Cahuzac, H.; Devel, L. Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices. Pharmaceuticals 2020, 13, 462. [Google Scholar] [CrossRef]
- Millet, A.; Khoudour, N.; Guitton, J.; Lebert, D.; Goldwasser, F.; Blanchet, B.; Machon, C. Analysis of Pembrolizumab in Human Plasma by LC-MS/HRMS. Method Valid. Comp. Elisa. Biomed. 2021, 9, 621. [Google Scholar] [CrossRef]
- Hallin, E.I.; Serkland, T.T.; Bjånes, T.K.; Skrede, S. High-throughput, low-cost quantification of 11 therapeutic antibodies using caprylic acid precipitation and LC-MS/MS. Anal. Chim. Acta 2024, 1313, 342789. [Google Scholar] [CrossRef]
- de Jong, K.A.; Rosing, H.; Huitema, A.D.; Beijnen, J.H. Optimized sample pre-treatment procedure for the simultaneous UPLC-MS/MS quantification of ipilimumab, nivolumab, and pembrolizumab in human serum. J. Chromatogr. B 2022, 1196, 123215. [Google Scholar] [CrossRef]
- Willeman, T.; Jourdil, J.-F.; Gautier-Veyret, E.; Bonaz, B.; Stanke-Labesque, F. A multiplex liquid chromatography tandem mass spectrometry method for the quantification of seven therapeutic monoclonal antibodies: Application for adalimumab therapeutic drug monitoring in patients with Crohn’s disease. Anal. Chim. Acta 2019, 1067, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Tron, C.; Lemaitre, F.; Bros, P.; Goulvestre, C.; Franck, B.; Mouton, N.; Bagnos, S.; Coriat, R.; Khoudour, N.; Lebert, D.; et al. Quantification of infliximab and adalimumab in human plasma by a liquid chromatography tandem mass spectrometry kit and comparison with two ELISA methods. Bioanalysis 2022, 14, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Scheffe, N.; Schreiner, R.; Thomann, A.; Findeisen, P. Development of a mass spectrometry-based method for quantification of ustekinumab in serum specimens. Ther. Drug Monit. 2020, 42, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Irie, K.; Hiramoto, N.; Hirabatake, M.; Ikesue, H.; Hashida, T.; Shimizu, T.; Ishikawa, T.; Muroi, N. Safety and blood levels of daratumumab after switching from intravenous to subcutaneous administration in patients with multiple myeloma. Investig. New Drugs 2023, 41, 761–767. [Google Scholar] [CrossRef]
- Li, W.; Huang, W.; Yu, X.; Chen, C.; Yuan, Y.; Liu, D.; Wang, F.; Yu, J.; Diao, X. A validated LC-MS/MS method for the quantitation of daratumumab in rat serum using rapid tryptic digestion without IgG purification and reduction. J. Pharm. Biomed. Anal. 2024, 243, 116083. [Google Scholar] [CrossRef]
- Abe, K.; Shibata, K.; Naito, T.; Karayama, M.; Hamada, E.; Maekawa, M.; Yamada, Y.; Suda, T.; Kawakami, J. Quantitative LC-MS/MS method for nivolumab in human serum using IgG purification and immobilized tryptic digestion. Anal. Methods 2020, 12, 54–62. [Google Scholar] [CrossRef]
- Millet, A.; Khoudour, N.; Bros, P.; Lebert, D.; Picard, G.; Machon, C.; Goldwasser, F.; Blanchet, B.; Guitton, J. Quantification of nivolumab in human plasma by LC-MS/HRMS and LC-MS/MS, comparison with ELISA. Talanta 2021, 224, 121889. [Google Scholar] [CrossRef]
- Heudi, O.; Barteau, S.; Zimmer, D.; Schmidt, J.; Bill, K.; Lehmann, N.; Bauer, C.; Kretz, O. Towards Absolute Quantification of Therapeutic Monoclonal Antibody in Serum by LC−MS/MS Using Isotope-Labeled Antibody Standard and Protein Cleavage Isotope Dilution Mass Spectrometry. Anal. Chem. 2008, 80, 4200–4207. [Google Scholar] [CrossRef]
- Wei, C.; Su, D.; Wang, J.; Jian, W.; Zhang, D. LC–MS challenges in characterizing and quantifying monoclonal antibodies (mAb) and antibody-drug conjugates (ADC) in biological samples. Curr. Pharmacol. Rep. 2018, 4, 45–63. [Google Scholar] [CrossRef]
- Hentschel, A.; Piontek, G.; Dahlmann, R.; Findeisen, P.; Sakson, R.; Carbow, P.; Renné, T.; Reinders, Y.; Sickmann, A. Highly sensitive therapeutic drug monitoring of infliximab in serum by targeted mass spectrometry in comparison to ELISA data. Clin. Proteom. 2024, 21, 16. [Google Scholar] [CrossRef]
- Xu, K.; Liu, L.; Maia, M.; Li, J.; Lowe, J.; Song, A.; Kaur, S. A multiplexed hybrid LC–MS/MS pharmacokinetic assay to measure two co-administered monoclonal antibodies in a clinical study. Bioanalysis 2014, 6, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-H.; Liao, H.-W.; Shao, Y.-Y.; Lu, Y.-S.; Lin, C.-H.; Tsai, I.-L.; Kuo, C.-H. Development of a general method for quantifying IgG-based therapeutic monoclonal antibodies in human plasma using protein G purification coupled with a two internal standard calibration strategy using LC-MS/MS. Anal. Chim. Acta 2018, 1019, 93–102. [Google Scholar] [CrossRef]
- Ouyang, Z.; Furlong, M.T.; Wu, S.; Sleczka, B.; Tamura, J.; Wang, H.; Suchard, S.; Suri, A.; Olah, T.; Tymiak, A. Pellet digestion: A simple and efficient sample preparation technique for LC–MS/MS quantification of large therapeutic proteins in plasma. Bioanalysis 2012, 4, 17–28. [Google Scholar] [CrossRef]
- Yang, Z.; Hayes, M.; Fang, X.; Daley, M.P.; Ettenberg, S.; Tse, F.L. LC− MS/MS Approach for Quantification of Therapeutic Proteins in Plasma Using a Protein Internal Standard and 2D-Solid-Phase Extraction Cleanup. Anal. Chem. 2007, 79, 9294–9301. [Google Scholar] [CrossRef]
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Bones, J. Multi-attribute method (MAM) to assess analytical comparability of adalimumab biosimilars. J. Pharm. Biomed. Anal. 2023, 234, 115543. [Google Scholar] [CrossRef]
- El Amrani, M.; Gerencser, L.; Huitema, A.D.; Hack, C.E.; van Luin, M.; van der Elst, K.C. A generic sample preparation method for the multiplex analysis of seven therapeutic monoclonal antibodies in human plasma or serum with liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2021, 1655, 462489. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, J.-Y.; Yang, W.; Zhang, H. Inhibition of protein carbamylation in urea solution using ammonium-containing buffers. Anal. Biochem. 2014, 446, 76–81. [Google Scholar] [CrossRef]
- Loo, R.R.O.; Dales, N.; Andrews, P.C. Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci. 1994, 3, 1975–1983. [Google Scholar] [CrossRef]
- Shieh, I.F.; Lee, C.-Y.; Shiea, J. Eliminating the Interferences from TRIS Buffer and SDS in Protein Analysis by Fused-Droplet Electrospray Ionization Mass Spectrometry. J. Proteome Res. 2005, 4, 606–612. [Google Scholar] [CrossRef]
- Rogers, J.C.; Bomgarden, R.D. Sample preparation for mass spectrometry-based proteomics; from proteomes to peptides. In Modern Proteomics—Sample Preparation, Analysis and Practical Applications; Springer: Berlin/Heidelberg, Germany, 2016; pp. 43–62. [Google Scholar]
- Lebert, D.; Picard, G.; Beau-Larvor, C.; Troncy, L.; Lacheny, C.; Maynadier, B.; Low, W.; Mouz, N.; Brun, V.; Klinguer-Hamour, C. Absolute and multiplex quantification of antibodies in serum using PSAQ™ standards and LC-MS/MS. Bioanalysis 2015, 7, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Martos, G.; Bedu, M.; Josephs, R.; Westwood, S.; Wielgosz, R. Quantification of SARS-CoV-2 monoclonal IgG mass fraction by isotope dilution mass spectrometry. Anal. Bioanal. Chem. 2024, 416, 2423–2437. [Google Scholar] [CrossRef] [PubMed]
- Suttapitugsakul, S.; Xiao, H.; Smeekens, J.; Wu, R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol. BioSyst. 2017, 13, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Winter, D. Systematic evaluation of protein reduction and alkylation reveals massive unspecific side effects by iodine-containing reagents. Mol. Cell. Proteom. 2017, 16, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, K.G.; Levitsky, L.I.; Pyatnitskiy, M.A.; Ilina, I.Y.; Bubis, J.A.; Solovyeva, E.M.; Zgoda, V.G.; Gorshkov, M.V.; Moshkovskii, S.A. Cysteine alkylation methods in shotgun proteomics and their possible effects on methionine residues. J. Proteom. 2021, 231, 104022. [Google Scholar] [CrossRef]
- Hains, P.G.; Robinson, P.J. The impact of commonly used alkylating agents on artifactual peptide modification. J. Proteome Res. 2017, 16, 3443–3447. [Google Scholar] [CrossRef]
- Geoghegan, K.F.; Hoth, L.R.; Tan, D.H.; Borzilleri, K.A.; Withka, J.M.; Boyd, J.G. Cyclization of N-terminal S-carbamoylmethylcysteine causing loss of 17 Da from peptides and extra peaks in peptide maps. J. Proteome Res. 2002, 1, 181–187. [Google Scholar] [CrossRef]
- Switzar, L.; Giera, M.; Niessen, W.M. Protein digestion: An overview of the available techniques and recent developments. J. Proteome Res. 2013, 12, 1067–1077. [Google Scholar] [CrossRef]
- Falck, D.; Jansen, B.C.; Plomp, R.; Reusch, D.; Haberger, M.; Wuhrer, M. Glycoforms of immunoglobulin G based biopharmaceuticals are differentially cleaved by trypsin due to the glycoform influence on higher-order structure. J. Proteome Res. 2015, 14, 4019–4028. [Google Scholar] [CrossRef]
- Solari, F.A.; Dell’Aica, M.; Sickmann, A.; Zahedi, R.P. Why phosphoproteomics is still a challenge. Mol. BioSystems 2015, 11, 1487–1493. [Google Scholar] [CrossRef]
- Heissel, S.; Frederiksen, S.J.; Bunkenborg, J.; Højrup, P. Enhanced trypsin on a budget: Stabilization, purification and high-temperature application of inexpensive commercial trypsin for proteomics applications. PLoS ONE 2019, 14, e0218374. [Google Scholar] [CrossRef] [PubMed]
- Menneteau, T.; Saveliev, S.; Butré, C.I.; Rivera, A.K.G.; Urh, M.; Delobel, A. Addressing common challenges of biotherapeutic protein peptide mapping using recombinant trypsin. J. Pharm. Biomed. Anal. 2024, 243, 116124. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Naito, T.; Okamura, J.; Hosokawa, S.; Mineta, H.; Kawakami, J. Simple and rapid LC-MS/MS method for the absolute determination of cetuximab in human serum using an immobilized trypsin. J. Pharm. Biomed. Anal. 2017, 146, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Reinders, L.M.; Klassen, M.D.; Teutenberg, T.; Jaeger, M.; Schmidt, T.C. Development of a multidimensional online method for the characterization and quantification of monoclonal antibodies using immobilized flow-through enzyme reactors. Anal. Bioanal. Chem. 2021, 413, 7119–7128. [Google Scholar] [CrossRef]
- Nagy, C.; Szabo, R.; Gaspar, A. Microfluidic immobilized enzymatic reactors for proteomic analyses—Recent developments and trends (2017–2021). Micromachines 2022, 13, 311. [Google Scholar] [CrossRef]
- Glatter, T.; Ludwig, C.; Ahrné, E.; Aebersold, R.; Heck, A.J.R.; Schmidt, A. Large-Scale Quantitative Assessment of Different In-Solution Protein Digestion Protocols Reveals Superior Cleavage Efficiency of Tandem Lys-C/Trypsin Proteolysis over Trypsin Digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Trevisiol, S.; Ayoub, D.; Lesur, A.; Ancheva, L.; Gallien, S.; Domon, B. The use of proteases complementary to trypsin to probe isoforms and modifications. Proteomics 2016, 16, 715–728. [Google Scholar] [CrossRef]
- Hao, P.; Ren, Y.; Datta, A.; Tam, J.P.; Sze, S.K. Evaluation of the effect of trypsin digestion buffers on artificial deamidation. J. Proteome Res. 2015, 14, 1308–1314. [Google Scholar] [CrossRef]
- Sutherland, E.; Veth, T.S.; Riley, N.M. Revisiting the effect of trypsin digestion buffers on artificial deamidation. ChemRxiv 2024. [Google Scholar] [CrossRef]
- Jiang, H.; Zeng, J.; Titsch, C.; Voronin, K.; Akinsanya, B.; Luo, L.; Shen, H.; Desai, D.D.; Allentoff, A.; Aubry, A.-F.; et al. Fully Validated LC-MS/MS Assay for the Simultaneous Quantitation of Coadministered Therapeutic Antibodies in Cynomolgus Monkey Serum. Anal. Chem. 2013, 85, 9859–9867. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Pipes, G.D.; Liu, D.; Shih, L.-Y.; Nichols, A.C.; Treuheit, M.J.; Brems, D.N.; Bondarenko, P.V. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal. Biochem. 2009, 392, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Neill, A.; Patel, R.; Andrien, B. Accurate determination of protein methionine oxidation by stable isotope labeling and LC-MS analysis. Anal. Chem. 2013, 85, 11705–11709. [Google Scholar] [CrossRef]
- Yang, J.; Gao, Z.; Ren, X.; Sheng, J.; Xu, P.; Chang, C.; Fu, Y. DeepDigest: Prediction of Protein Proteolytic Digestion with Deep Learning. Anal. Chem. 2021, 93, 6094–6103. [Google Scholar] [CrossRef]
- Gupta, S.; Jiskoot, W.; Schöneich, C.; Rathore, A.S. Oxidation and deamidation of monoclonal antibody products: Potential impact on stability, biological activity, and efficacy. J. Pharm. Sci. 2022, 111, 903–918. [Google Scholar] [CrossRef]
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Buchanan, T.; Guender, M.; Kristensen, D.B.; Sloth, T.M.; Ørgaard, M.; Cook, K.; Bones, J. Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Anal. Bioanal. Chem. 2020, 412, 6833–6848. [Google Scholar] [CrossRef]
- Kuzyk, M.A.; Parker, C.E.; Domanski, D.; Borchers, C.H. Development of MRM-based assays for the absolute quantitation of plasma proteins. Methods Mol. Biol. 2013, 1023, 53–82. [Google Scholar] [CrossRef]
- Liebler, D.C.; Zimmerman, L.J. Targeted quantitation of proteins by mass spectrometry. Biochemistry 2013, 52, 3797–3806. [Google Scholar] [CrossRef]
- Schmidt, C.; Urlaub, H. Absolute quantification of proteins using standard peptides and multiple reaction monitoring. Quant. Methods Proteom. 2012, 893, 249–265. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. 2002, 310–322. [Google Scholar] [CrossRef]
- Pugalenthi, G.; Nithya, V.; Chou, K.-C.; Archunan, G. Nglyc: A random forest method for prediction of N-glycosylation sites in eukaryotic protein sequence. Protein Pept. Lett. 2020, 27, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.C.; Aoki-Kinoshita, K.F.; Caragea, D.; Kc, D.B. DeepNGlyPred: A deep neural network-based approach for human N-linked glycosylation site prediction. Molecules 2021, 26, 7314. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Kang, L.; Burton, L.; Weng, N. A workflow for absolute quantitation of large therapeutic proteins in biological samples at intact level using LC-HRMS. Bioanalysis 2016, 8, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Lanshoeft, C.; Cianférani, S.; Heudi, O. Generic Hybrid Ligand Binding Assay Liquid Chromatography High-Resolution Mass Spectrometry-Based Workflow for Multiplexed Human Immunoglobulin G1 Quantification at the Intact Protein Level: Application to Preclinical Pharmacokinetic Studies. Anal. Chem. 2017, 89, 2628–2635. [Google Scholar] [CrossRef]
- Cong, Y.; Zhang, Z.; Zhang, S.; Hu, L.; Gu, J. Quantitative MS analysis of therapeutic mAbs and their glycosylation for pharmacokinetics study. Proteom.—Clin. Appl. 2016, 10, 303–314. [Google Scholar] [CrossRef]
- Liu, S.; Liu, X. IgG N-glycans. Adv. Clin. Chem. 2021, 105, 1–47. [Google Scholar] [CrossRef]
- van den Broek, I.; van Dongen, W.D. LC–MS-based quantification of intact proteins: Perspective for clinical and bioanalytical applications. Bioanalysis 2015, 7, 1943–1958. [Google Scholar] [CrossRef]
- Rosati, S.; Yang, Y.; Barendregt, A.; Heck, A.J.R. Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nat. Protoc. 2014, 9, 967–976. [Google Scholar] [CrossRef]
- Xu, K.; Liu, L.; Saad, O.M.; Baudys, J.; Williams, L.; Leipold, D.; Shen, B.; Raab, H.; Junutula, J.R.; Kim, A. Characterization of intact antibody–drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography–mass spectrometry. Anal. Biochem. 2011, 412, 56–66. [Google Scholar] [CrossRef]
- Jin, W.; Burton, L.; Moore, I. LC–HRMS quantitation of intact antibody drug conjugate trastuzumab emtansine from rat plasma. Bioanalysis 2018, 10, 851–862. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, H.; Zheng, N.; Zeng, J. Critical considerations for immunocapture enrichment LC–MS bioanalysis of protein therapeutics and biomarkers. Bioanalysis 2018, 10, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Weng, N.; Jian, W. LC–MS bioanalysis of intact proteins and peptides. Biomed. Chromatogr. 2020, 34, e4633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Vasicek, L.A.; Hsieh, S.; Zhang, S.; Bateman, K.P.; Henion, J. Top-down LC–MS quantitation of intact denatured and native monoclonal antibodies in biological samples. Bioanalysis 2018, 10, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.E.; Lebert, D.; Guillaubez, J.-V.; Samra, S.; Goucher, E.; Hart, B. Streamlined Workflow for Absolute Quantitation of Therapeutic Monoclonal Antibodies Using Promise Proteomics mAbXmise Kits and a TSQ Altis Plus Mass Spectrometer. Available online: https://promise-proteomics.com/wp-content/uploads/2023/06/tn-001753-cl-clinical-altis-mabs-tn001753-na-en.pdf (accessed on 7 August 2024).
- Rutherfurd, S.M.; Gilani, G.S. Amino Acid Analysis. Curr. Protoc. Protein Sci. 2009, 58, 11.9.1–11.9.37. [Google Scholar] [CrossRef]
- Rutherfurd, S.M.; Dunn, B.M. Quantitative Amino Acid Analysis. Curr. Protoc. Protein Sci. 2011, 63, 3.2.1–3.2.6. [Google Scholar] [CrossRef]
- Josephs, R.D.; Martos, G.; Li, M.; Wu, L.; Melanson, J.E.; Quaglia, M.; Beltrão, P.J.; Prevoo-Franzsen, D.; Boeuf, A.; Delatour, V. Establishment of measurement traceability for peptide and protein quantification through rigorous purity assessment—A review. Metrologia 2019, 56, 044006. [Google Scholar] [CrossRef]
- Violi, J.P.; Bishop, D.P.; Padula, M.P.; Steele, J.R.; Rodgers, K.J. Considerations for amino acid analysis by liquid chromatography-tandem mass spectrometry: A tutorial review. TrAC Trends Anal. Chem. 2020, 131, 116018. [Google Scholar] [CrossRef]
- Liu, F.; Lai, S.; Tong, H.; Lakey, P.S.; Shiraiwa, M.; Weller, M.G.; Pöschl, U.; Kampf, C.J. Release of free amino acids upon oxidation of peptides and proteins by hydroxyl radicals. Anal. Bioanal. Chem. 2017, 409, 2411–2420. [Google Scholar] [CrossRef]
- Kato, M.; Kato, H.; Eyama, S.; Takatsu, A. Application of amino acid analysis using hydrophilic interaction liquid chromatography coupled with isotope dilution mass spectrometry for peptide and protein quantification. J. Chromatogr. B 2009, 877, 3059–3064. [Google Scholar] [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; Schrag, J.D.; Melanson, J.E. Characterization of a SARS-CoV-2 spike protein reference material. Anal. Bioanal. Chem. 2022, 414, 3561–3569. [Google Scholar] [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; L’Abbé, D.; Stuible, M.; Durocher, Y.; Melanson, J.E. Production and Characterization of a SARS-CoV-2 Nucleocapsid Protein Reference Material. ACS Meas. Sci. 2022, 2, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Louwagie, M.; Kieffer-Jaquinod, S.; Dupierris, V.; Couté, Y.; Bruley, C.; Garin, J.; Dupuis, A.; Jaquinod, M.; Brun, V. Introducing AAA-MS, a Rapid and Sensitive Method for Amino Acid Analysis Using Isotope Dilution and High-Resolution Mass Spectrometry. J. Proteome Res. 2012, 11, 3929–3936. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-S.; Lim, H.-M.; Kim, S.-K.; Ku, H.-K.; Oh, K.-H.; Park, S.-R. Quantification of human growth hormone by amino acid composition analysis using isotope dilution liquid-chromatography tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 6596–6602. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Josephs, R.; Melanson, J.; Dai, X.; Wang, Y.; Zhai, R.; Chu, Z.; Fang, X.; Thibeault, M.; Stocks, B. PAWG Pilot Study on Quantification of SARS-CoV-2 Monoclonal Antibody—Part 1. Metrologia 2022, 59, 08001. [Google Scholar] [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; L’Abbé, D.; Umer, M.; Liu, Y.; Stuible, M.; Durocher, Y.; Melanson, J.E. Characterization of biotinylated human ACE2 and SARS-CoV-2 Omicron BA. 4/5 spike protein reference materials. Anal. Bioanal. Chem. 2024, 416, 4861–4872. [Google Scholar] [CrossRef]
- Catalogue: SIL-mAbs for Targeted LC-MS Quantification. Available online: https://promise-proteomics.com/wp-content/uploads/2024/01/Catalogue-SIL-mAbs-2024.pdf (accessed on 7 August 2024).
- Picard, G.; Lebert, D.; Louwagie, M.; Adrait, A.; Huillet, C.; Vandenesch, F.; Bruley, C.; Garin, J.; Jaquinod, M.; Brun, V. PSAQ™ standards for accurate MS-based quantification of proteins: From the concept to biomedical applications. J. Mass Spectrom. 2012, 47, 1353–1363. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Osaki, F.; Tabata, K.; Oe, T. Quantitative LC/ESI-SRM/MS of antibody biopharmaceuticals: Use of a homologous antibody as an internal standard and three-step method development. Anal. Bioanal. Chem. 2017, 409, 5523–5532. [Google Scholar] [CrossRef]
- Smit, N.P.M.; Ruhaak, L.R.; Romijn, F.P.H.T.M.; Pieterse, M.M.; Van Der Burgt, Y.E.M.; Cobbaert, C.M. The Time Has Come for Quantitative Protein Mass Spectrometry Tests That Target Unmet Clinical Needs. J. Am. Soc. Mass Spectrom. 2021, 32, 636–647. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J. Peptide purity assignment for antibody quantification by combining isotope dilution mass spectrometry and liquid chromatography. Bull. Korean Chem. Soc. 2022, 43, 704–713. [Google Scholar] [CrossRef]
- Burkitt, W.I.; Pritchard, C.; Arsene, C.; Henrion, A.; Bunk, D.; O’Connor, G. Toward Systeme International d’Unite-traceable protein quantification: From amino acids to proteins. Anal. Biochem. 2008, 376, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Benesova, E.; Vidova, V.; Spacil, Z. A comparative study of synthetic winged peptides for absolute protein quantification. Sci. Rep. 2021, 11, 10880. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ortiz, R.; Tran, L.; Hall, M.; Spahr, C.; Walker, K.; Laudemann, J.; Miller, S.; Salimi-Moosavi, H.; Lee, J.W. General LC-MS/MS Method Approach to Quantify Therapeutic Monoclonal Antibodies Using a Common Whole Antibody Internal Standard with Application to Preclinical Studies. Anal. Chem. 2012, 84, 1267–1273. [Google Scholar] [CrossRef]
- Bronsema, K.J.; Bischoff, R.; van de Merbel, N.C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr. B 2012, 893, 1–14. [Google Scholar] [CrossRef]
- Furlong, M.T.; Ouyang, Z.; Wu, S.; Tamura, J.; Olah, T.; Tymiak, A.; Jemal, M. A universal surrogate peptide to enable LC-MS/MS bioanalysis of a diversity of human monoclonal antibody and human Fc-fusion protein drug candidates in pre-clinical animal studies. Biomed. Chromatogr. 2012, 26, 1024–1032. [Google Scholar] [CrossRef]
- Furlong, M.T. Generic Peptide Strategies for LC–MS/MS Bioanalysis of Human Monoclonal Antibody Drugs and Drug Candidates. In Protein Analysis Using Mass Spectrometry: Accelerating Protein Biotherapeutics from Lab to Patient; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 161–181. [Google Scholar]
- Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 7 August 2024).
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on 7 August 2024).
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef]
- Ranbaduge, N.; Yu, Y.Q. A Streamlined Compliant Ready Workflow for Peptide-Based Multi-Attribute Method (MAM). Available online: https://www.waters.com/content/dam/waters/en/app-notes/2020/720007094/720007094-zh_tw.pdf (accessed on 7 August 2024).
- Yun, W.; Alelyunas, H.S.; Wrona, M.D.; Chen, W. Rapid, Sensitive, and Routine Intact mAb Quantification Using a Compact Tof HRMS Platform. Available online: https://lcms.cz/labrulez-bucket-strapi-h3hsga3/2019asms_alelyunas_intactproteinquan_659b6f83aa/2019asms_alelyunas_intactproteinquan.pdf (accessed on 7 August 2024).
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Bones, J. Multi-Attribute Method (MAM) Analytical Workflow for Biotherapeutic Protein Characterization from Process Development to QC. Curr. Protoc. 2023, 3, e927. [Google Scholar] [CrossRef]
- Schneck, N.A.; Mehl, J.T.; Kellie, J.F. Protein LC-MS Tools for the Next Generation of Biotherapeutic Analyses from Preclinical and Clinical Serum. J. Am. Soc. Mass Spectrom. 2023, 34, 1837–1846. [Google Scholar] [CrossRef]
- Kiyonami, R.; Schoen, A.; Prakash, A.; Nguyen, H.; Peterman, S.; Selevsek, N.; Zabrouskov, V.; Huhmer, A.; Domon, B. Rapid Assay Development and Refinement for Targeted Protein Quantitation Using an Intelligent SRM (iSRM) Workflow. Available online: https://tools.thermofisher.com/content/sfs/brochures/AN468_63139_Vantage_Prot(1).pdf (accessed on 7 August 2024).
- Kiyonami, R.; Zeller, M.; Zabrouskov, V. Quantifying Peptides in Complex Mixtures with High Sensitivity and Precision Using a Targeted Approach with a Hybrid Linear Ion Trap-Orbitrap Mass Spectrometer. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-557-LC-MS-Peptides-Complex-Mixtures-AN63499-EN.pdf (accessed on 7 August 2024).
- Colangelo, C.M.; Chung, L.; Bruce, C.; Cheung, K.-H. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods 2013, 61, 287–298. [Google Scholar] [CrossRef]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Pfeuffer, J.; Bielow, C.; Wein, S.; Jeong, K.; Netz, E.; Walter, A.; Alka, O.; Nilse, L.; Colaianni, P.D.; McCloskey, D. OpenMS 3 enables reproducible analysis of large-scale mass spectrometry data. Nat. Methods 2024, 21, 365–367. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kanazawa, M.; Ogiwara, A.; Arita, M. MRMPROBS suite for metabolomics using large-scale MRM assays. Bioinformatics 2014, 30, 2379–2380. [Google Scholar] [CrossRef]
- Valot, B.; Langella, O.; Nano, E.; Zivy, M. MassChroQ: A versatile tool for mass spectrometry quantification. Proteomics 2011, 11, 3572–3577. [Google Scholar] [CrossRef]
- Cai, Y.; Weng, K.; Guo, Y.; Peng, J.; Zhu, Z.-J. An integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing. Metabolomics 2015, 11, 1575–1586. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 7 August 2024).
- Choi, M.; Carver, J.; Chiva, C.; Tzouros, M.; Huang, T.; Tsai, T.-H.; Pullman, B.; Bernhardt, O.M.; Hüttenhain, R.; Teo, G.C. MassIVE. quant: A community resource of quantitative mass spectrometry–based proteomics datasets. Nat. Methods 2020, 17, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Pfeuffer, J.; Wang, H.; Zheng, P.; Käll, L.; Sachsenberg, T.; Demichev, V.; Bai, M.; Kohlbacher, O.; Perez-Riverol, Y. quantms: A cloud-based pipeline for quantitative proteomics enables the reanalysis of public proteomics data. Nat. Methods 2024, 21, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiang, H.; Lu, M.; Tong, J.; An, S.; Wang, J.; Yu, C. MRMPro: A web-based tool to improve the speed of manual calibration for multiple reaction monitoring data analysis by mass spectrometry. BMC Bioinform. 2024, 25, 60. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, C.; Zeng, W.-F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Mi, W.; Josephs, R.; Melanson, J.; Dai, X.; Wang, Y.; Zhai, R.; Chu, Z.; Fang, X.; Thibeault, M.; Stocks, B. PAWG pilot study on quantification of SARS-CoV-2 monoclonal antibody—Part 2. Metrologia 2023, 60, 08016. [Google Scholar] [CrossRef]
- Miller, W.G.; Greenberg, N.; Panteghini, M.; Budd, J.R.; Johansen, J.V. Guidance on which calibrators in a metrologically traceable calibration hierarchy must be commutable with clinical samples. Clin. Chem. 2023, 69, 228–238. [Google Scholar] [CrossRef]
- Diederiks, N.M.; van der Burgt, Y.E.; Ruhaak, L.R.; Cobbaert, C.M. Developing an SI-traceable Lp (a) reference measurement system: A pilgrimage to selective and accurate apo (a) quantification. Crit. Rev. Clin. Lab. Sci. 2023, 60, 483–501. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Romijn, F.P.; Begcevic Brkovic, I.; Kuklenyik, Z.; Dittrich, J.; Ceglarek, U.; Hoofnagle, A.N.; Althaus, H.; Angles-Cano, E.; Coassin, S. Development of an LC-MRM-MS-based candidate reference measurement procedure for standardization of serum apolipoprotein (a) tests. Clin. Chem. 2023, 69, 251–261. [Google Scholar] [CrossRef]
- Schiel, J.E.; Mire-Sluis, A.; Davis, D. Monoclonal antibody therapeutics: The need for biopharmaceutical reference materials. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space; ACS Publications: Washington, DC, USA, 2014; pp. 1–34. [Google Scholar]
- Schiel, J.E.; Turner, A. The NISTmAb Reference Material 8671 lifecycle management and quality plan. Anal. Bioanal. Chem. 2018, 410, 2067–2078. [Google Scholar] [CrossRef]
- Kinumi, T.; Saikusa, K.; Kato, M.; Kojima, R.; Igarashi, C.; Noda, N.; Honda, S. Characterization and Value Assignment of a Monoclonal Antibody Reference Material, NMIJ RM 6208a, AIST-MAB. Front. Mol. Biosci. 2022, 9, 842041. [Google Scholar] [CrossRef]
- Dong, L.; Zhang, Y.; Fu, B.; Swart, C.; Jiang, H.; Liu, Y.; Huggett, J.; Wielgosz, R.; Niu, C.; Li, Q. Reliable biological and multi-omics research through biometrology. Anal. Bioanal. Chem. 2024, 416, 3645–3663. [Google Scholar] [CrossRef] [PubMed]
- Shuford, C.M.; Walters, J.J.; Holland, P.M.; Sreenivasan, U.; Askari, N.; Ray, K.; Grant, R.P. Absolute protein quantification by mass spectrometry: Not as simple as advertised. Anal. Chem. 2017, 89, 7406–7415. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chen, H.; Zare, R.N. Ultrafast enzymatic digestion of proteins by microdroplet mass spectrometry. Nat. Commun. 2020, 11, 1049. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Li, Z.; Xing, X.; He, F.; Huang, J.; Xue, D. Improving the activity and thermal stability of trypsin by the rational design. Process Biochem. 2023, 130, 227–235. [Google Scholar] [CrossRef]
- Guo, C.; Liu, Y.; Yu, H.; Du, K.; Gan, Y.; Huang, H. A novel strategy for thermostability improvement of trypsin based on N-glycosylation within the Ω-loop region. J. Microbiol. Biotechnol. 2016, 26, 1163–1172. [Google Scholar] [CrossRef]
- Narumi, R.; Masuda, K.; Tomonaga, T.; Adachi, J.; Ueda, H.R.; Shimizu, Y. Cell-free synthesis of stable isotope-labeled internal standards for targeted quantitative proteomics. Synth. Syst. Biotechnol. 2018, 3, 97–104. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Antibody Subclass | Year of Approval | Target | Sales 1 (Billion USD) | Signature Peptides 2 |
|---|---|---|---|---|---|
| Pembrolizumab | IgG4κ | 2021 | Programmed cell death protein 1 (PD-1) | 25.0 | ASGYTFTNYYMYWVR [28] DLPLTFGGGTK [28,29] VTLTTDSSTTTAYMELK [30] |
| Adalimumab | IgG1κ | 2002 | Tumor necrosis factor alpha (TNF-α) | 14.4 | APYTFGQGTK [31,32] |
| Ustekinumab | IgG1κ | 2009 | Interleukin (IL)-12 and IL-23 | 10.9 | PGQGYFDFWGQGTLVTVSSSSTK [33] GLDWIGIMSPVDSDIR [29,33] |
| Daratumumab | IgG1κ | 2016 | Hydrolase CD38 | 9.7 | SNWPPTFGQGTK [34] LLIYDASNR [35] |
| Nivolumab | IgG4κ | 2014 | PD-1 | 9.0 | ASGITFSNSGMHWVR [29,30,36,37] |
| Product | Special Feature | Denaturation | Reduction | Alkylation | Digestion Conditions | Ref. |
|---|---|---|---|---|---|---|
| Trypsin | ||||||
| Promega GmbH (Walldorf, BW, GER) | ||||||
| Trypsin Gold/Sequencing Grade | Maximum specificity | 8 M Urea 1 h | DTT | IAM 30 min | Overnight 37 °C | [28,31,37] |
| Trypsin Platinum | Recombinant enzyme, autoproteolytic resistance | 8 M GuHCl 30 min | TCEP | IAM 30 min | Overnight 37 °C | |
| Thermo Fisher Scientific (Waltham, MA, USA) | ||||||
| Pierce™ Trypsin | 1 h at 60 °C or 10 min at 95 °C | DTT | IAA, 30 min | 4 to 24 h 37 °C | [29,30] | |
| SMART Digest Trypsin-Kit | Automatable process | - | opt. | opt. | 45 min (IgG) 70 °C | |
| In-Solution Tryptic Digestion and Guanidination Kit | Improved ionization by guanidination of K into homo-R | 95 °C 5 min | DTT | IAM, 30 min | 2 h at 37 °C or overnight at 30 °C | |
| Waters Corporation (Milford, MA, USA) | ||||||
| ProteinWorks eXpress Digest Kit | High throughput of samples possible | Digestion buffer, 80 °C, 10 min | Reduction agent 60 °C, 20 min | Alkylation agent 30 min | 2 h 45 °C | [33] |
| Promise Proteomics (Grenoble, ARA, FRA) | ||||||
| mAbXmise Kit | Immunocapture cartridges | opt., 4 M to 0.1 M Urea | - | - | 30 min to 15 h 37 °C | [32] |
| Trypsin/ Lys-C Mix | ||||||
| Thermo Fisher Scientific (Waltham, MA, USA) | ||||||
| EasyPep™ Mini MS Sample Prep Kit | High throughput of samples possible | Lysis solution 95 °C, 10 min | Red. Solution | Alk. Solution | 1 to 3 h 37 °C | |
| Pierce™ Trypsin/ Lys-C Protease Mix | 8 M Urea, 1 h at 60 °C or 10 min at 95 °C | DTT | IAM, 30 min | 2 to 16 h 37 °C | ||
| Promega GmbH (Walldorf, BW, GER) | ||||||
| Rapid Digestion– Trypsin/LysC | Fast digestion | - | opt. | opt. | 1 h 70 °C | |
| Trypsin/Lys-C | Quantification | 6–8 M Urea, 30 min | DTT | IAM, 30 min | overnight 37 °C | |
| Manufacturer/Lab | Software |
|---|---|
| Waters Coporation (Milford, MA, USA) | BioAccord System (UNIFI software, version 1.9.9), TargetLynx™ and QuanOptimize™ (integrated in MassLynx version 4.2) |
| Thermo Fisher Scientific (Waltham, MA, USA) | Xcalibur (version 4.3), BioPharma FinderTM (version 5.3) and PinpointTM (version 4.1) |
| Agilent Technologies (Santa Clara, CA, USA) | MassHunter (version 12.0) |
| AB Sciex (Framingham, MA, USA) | Analyst® (version 1.7.3), MultiQuantTM (version 3.0.3) and MRMPilotTM (version 2.1) |
| Biognosys AG (Schlieren, DIE, ZH) | SpectronautTM (version 19), SpectromineTM (version 3), and SpectroDiveTM (version 12) |
| MacCoss Lab (Seattle, WA, USA) | Skyline (version 24.1) 1 |
| Cox Lab (Martinsried, BY, GER) | MaxQuant (version 2.6.7.0) 1,2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Döring, S.; Weller, M.G.; Reinders, Y.; Konthur, Z.; Jaeger, C. Challenges and Insights in Absolute Quantification of Recombinant Therapeutic Antibodies by Mass Spectrometry: An Introductory Review. Antibodies 2025, 14, 3. https://doi.org/10.3390/antib14010003
Döring S, Weller MG, Reinders Y, Konthur Z, Jaeger C. Challenges and Insights in Absolute Quantification of Recombinant Therapeutic Antibodies by Mass Spectrometry: An Introductory Review. Antibodies. 2025; 14(1):3. https://doi.org/10.3390/antib14010003
Chicago/Turabian StyleDöring, Sarah, Michael G. Weller, Yvonne Reinders, Zoltán Konthur, and Carsten Jaeger. 2025. "Challenges and Insights in Absolute Quantification of Recombinant Therapeutic Antibodies by Mass Spectrometry: An Introductory Review" Antibodies 14, no. 1: 3. https://doi.org/10.3390/antib14010003
APA StyleDöring, S., Weller, M. G., Reinders, Y., Konthur, Z., & Jaeger, C. (2025). Challenges and Insights in Absolute Quantification of Recombinant Therapeutic Antibodies by Mass Spectrometry: An Introductory Review. Antibodies, 14(1), 3. https://doi.org/10.3390/antib14010003