Impact of Monoclonal Antibody Aggregates on Effector Function Characterization

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Forced Degradation Study
2.3. Photostability Studies
2.4. Size Exclusion Chromatography (SEC)
2.4.1. Analytical SEC
2.4.2. Preparative SEC
2.5. Capillary Gel Electrophoresis–Sodium Dodecyl Sulfate (CE-SDS)
2.6. Mass Spectrometry
2.7. SPR Binding Assays with Ref Standard Curves
2.8. Kinetics, Affinity, and Direct Binding SPR Assays
2.9. Solution-Based Competitive Fc Receptor Binding Assays
2.10. FcγRIIa and FcγRIIIa Reporter Gene Assays
2.10.1. FcγRIIa Assay
2.10.2. FcγRIIIa Assay
2.11. Statistical Data Analysis
3. Results
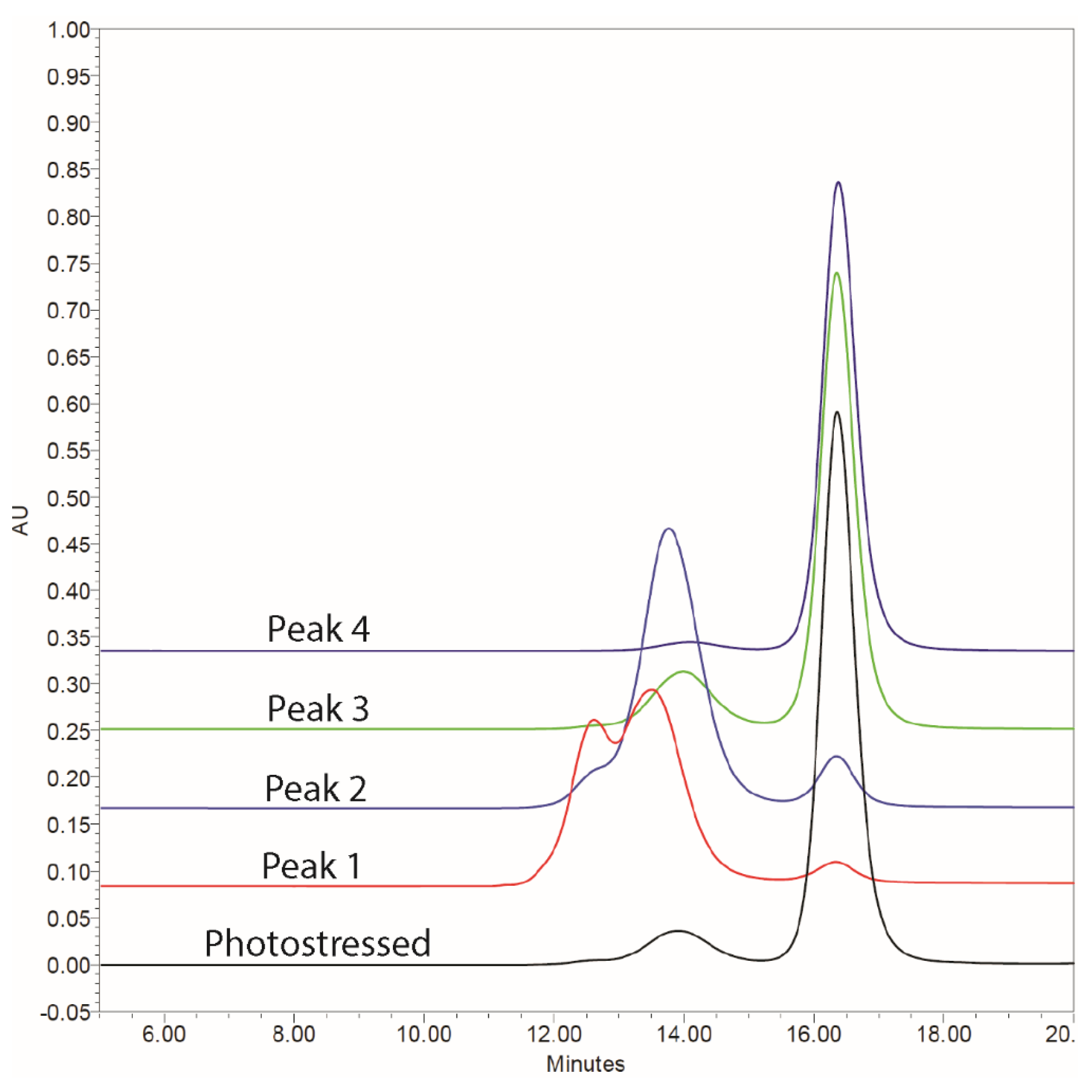
3.1. Characterization of Photostability and Forcibly Degraded mAb
3.2. Activity of Isolated Aggregate mAb Fractions
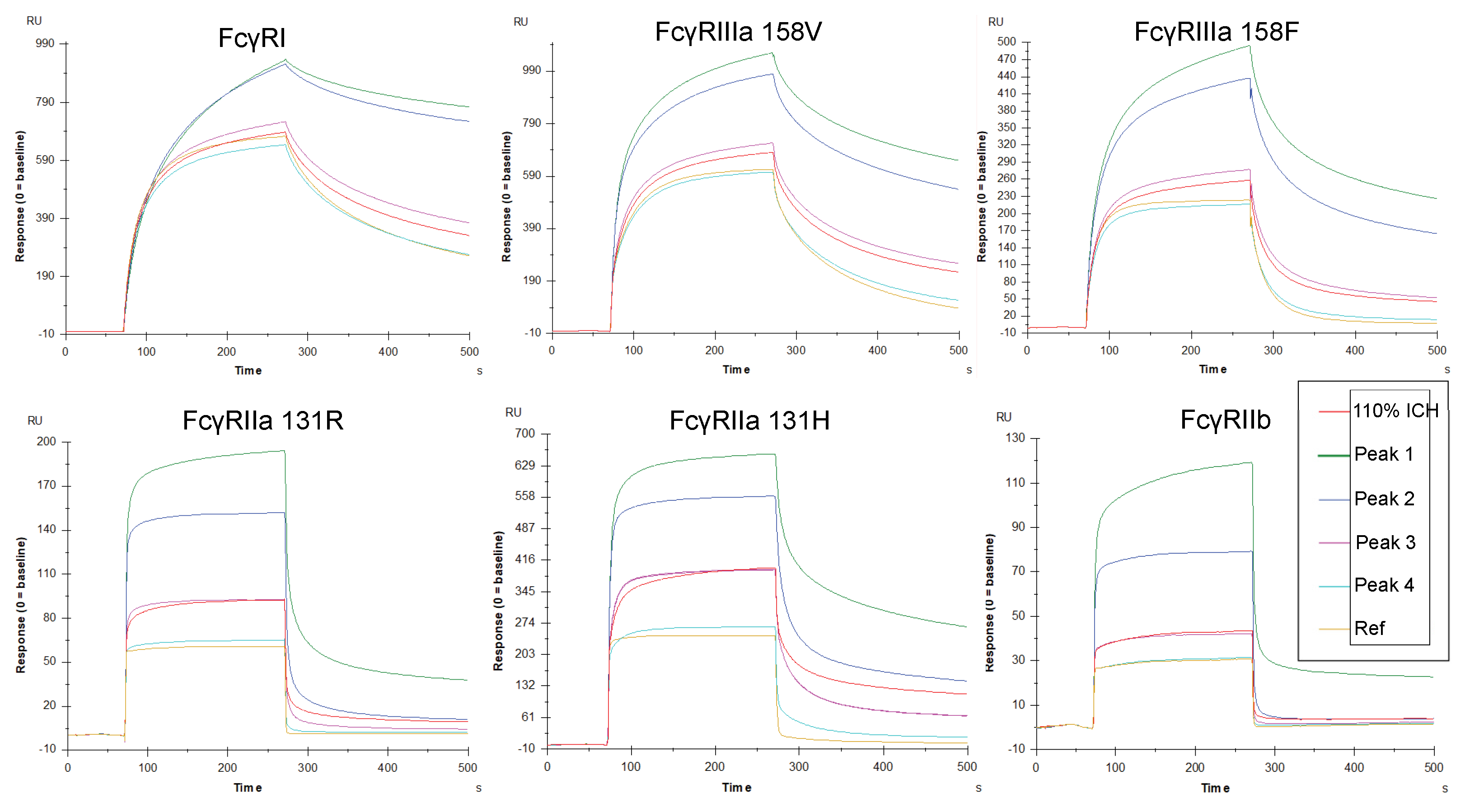
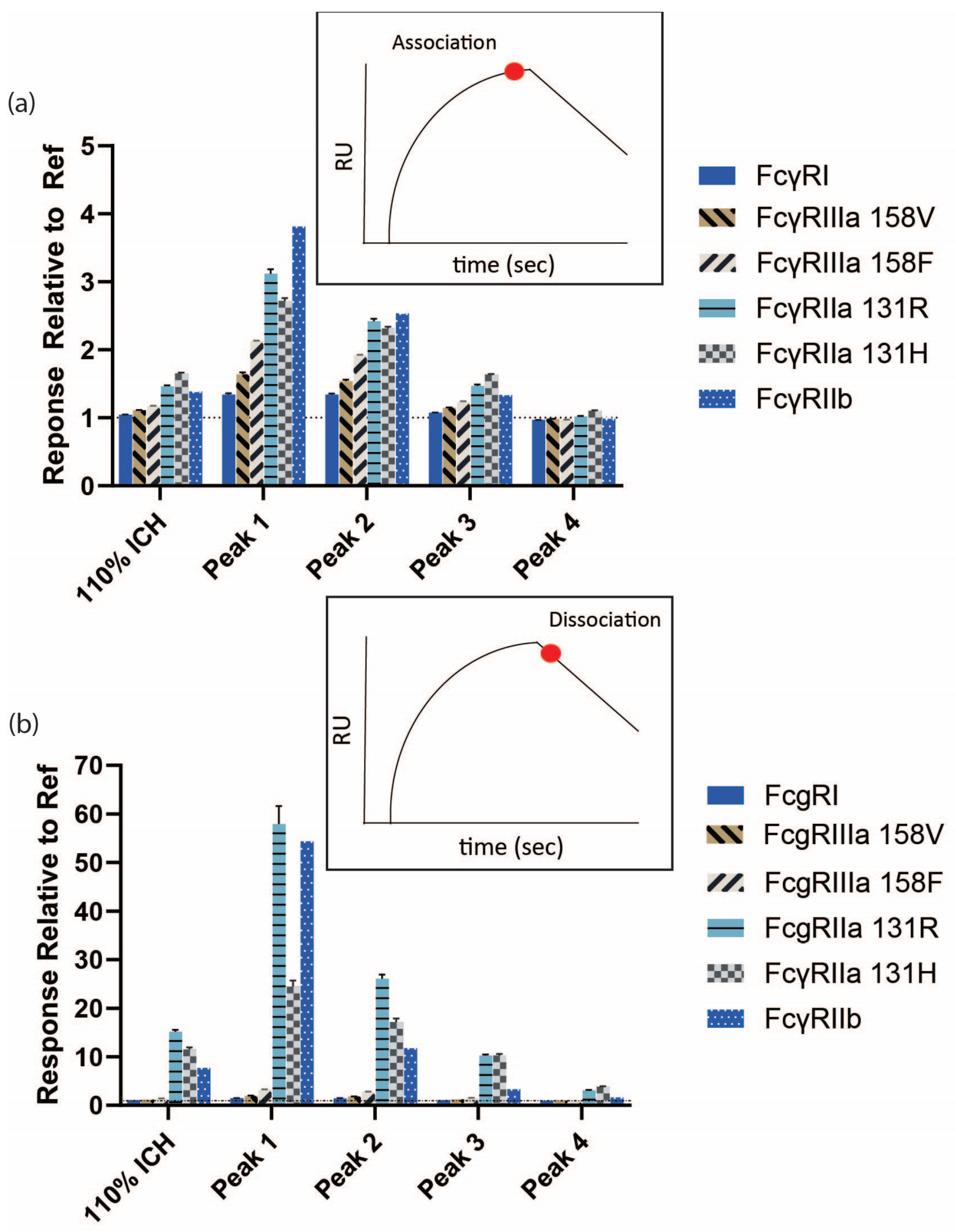
3.3. Increased Fc Receptor Binding by SPR Is Avidity-Driven
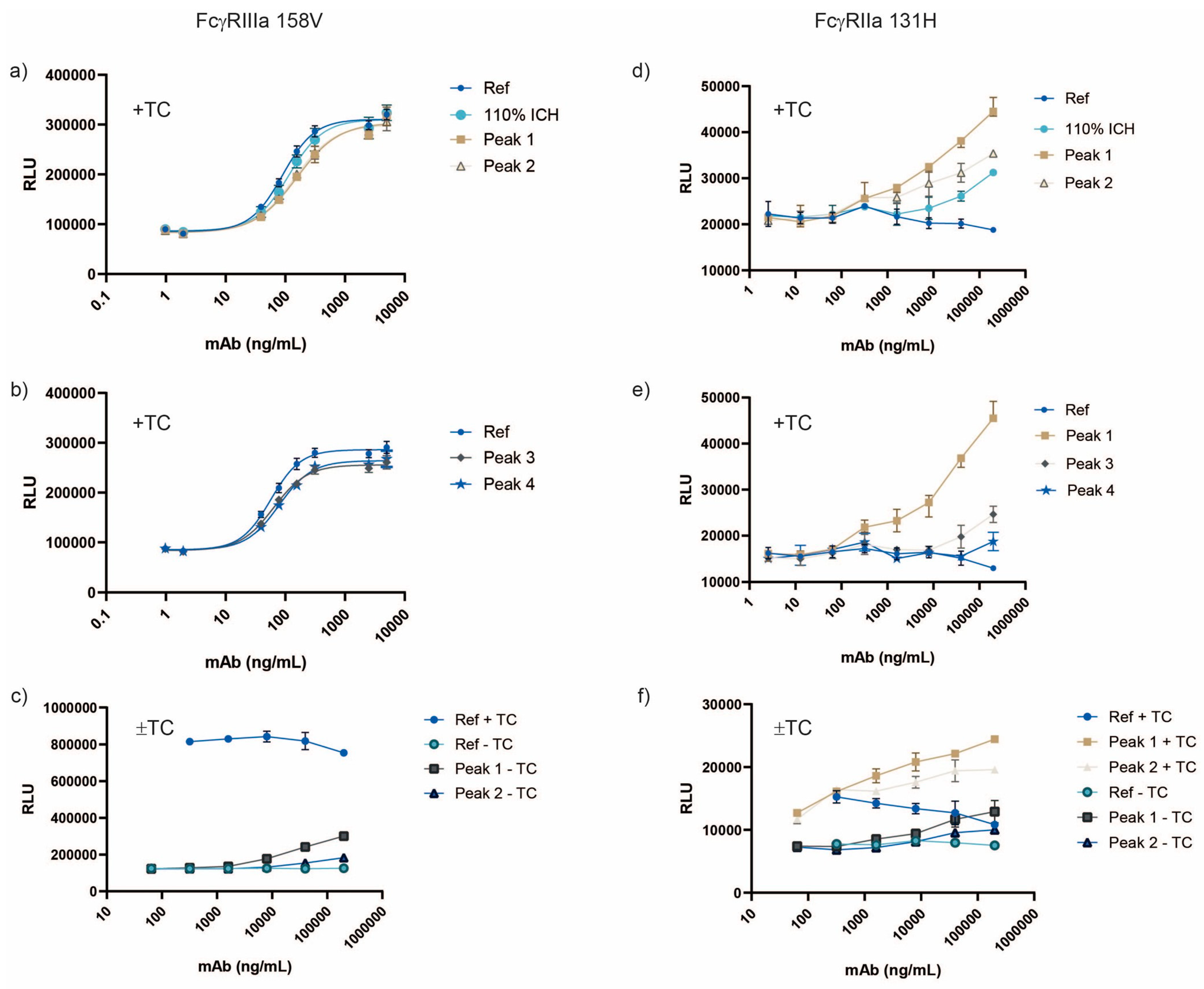
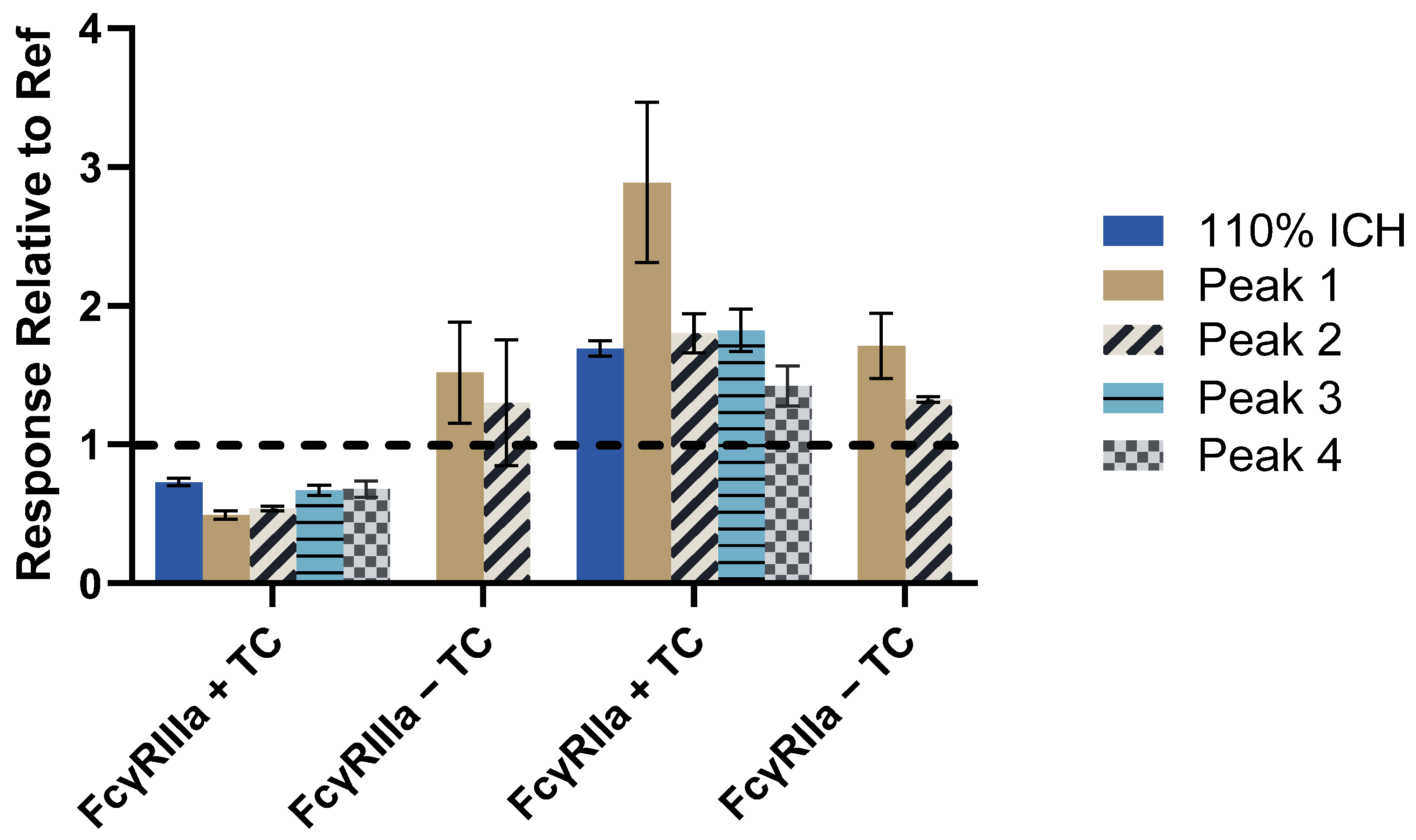
3.4. Aggregation Increases FcγRIIa Activity More than Other Fc Receptors in Various Assay Formats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | anti-drug antibody |
| ADCC | antibody-dependent cellular cytotoxicity |
| ADCP | antibody-dependent cellular phagocytosis |
| CE-SDS | capillary electrophoresis sodium dodecyl sulfate |
| FcRn | neonatal Fc receptor |
| mAb | monoclonal antibody |
| IVIG | intravenous immunoglobulin |
| PBMC | peripheral blood mononuclear cells |
| Ref | one mAb1 reference standard batch used throughout the study |
| RU | resonance units |
| RLU | relative luminescence units |
| SA | streptavidin |
| SEC | size exclusion chromatography |
| SPR | surface plasmon resonance |
References
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar] [PubMed]
- Patel, K.R.; Roberts, J.T.; Barb, A.W. Multiple Variables at the Leukocyte Cell Surface Impact Fc gamma Receptor-Dependent Mechanisms. Front. Immunol. 2019, 10, 223. [Google Scholar]
- Rus, H.; Cudrici, C.; Niculescu, F. The Role of the Complement System in Innate Immunity. Immunol. Res. 2005, 33, 103–112. [Google Scholar]
- Kim, J.; Lee, J.Y.; Kim, H.G.; Kwak, M.W.; Kang, T.H. Fc Receptor Variants and Disease: A Crucial Factor to Consider in the Antibody Therapeutics in Clinic. Int. J. Mol. Sci. 2021, 22, 9489. [Google Scholar] [CrossRef]
- Herter, S.; Birk, M.C.; Klein, C.; Gerdes, C.; Umana, P.; Bacac, M. Glycoengineering of therapeutic antibodies enhances monocyte/macrophage-mediated phagocytosis and cytotoxicity. J. Immunol. 2014, 192, 2252–2260. [Google Scholar]
- Kerntke, C.; Nimmerjahn, F.; Biburger, M. There Is (Scientific) Strength in Numbers: A Comprehensive Quantitation of Fc Gamma Receptor Numbers on Human and Murine Peripheral Blood Leukocytes. Front. Immunol. 2020, 11, 118. [Google Scholar]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar]
- Lundahl, M.L.E.; Fogli, S.; Colavita, P.E.; Scanlan, E.M. Aggregation of protein therapeutics enhances their immunogenicity: Causes and mitigation strategies. RSC Chem. Biol. 2021, 2, 1004–1020. [Google Scholar]
- Wen, Y.; Jawa, V. The Impact of Product and Process Related Critical Quality Attributes on Immunogenicity and Adverse Immunological Effects of Biotherapeutics. J. Pharm. Sci. 2021, 110, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Effects of Protein Aggregates: An Immunologic Perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef]
- Bansal, R.; Gupta, S.; Rathore, A.S. Analytical Platform for Monitoring Aggregation of Monoclonal Antibody Therapeutics. Pharm. Res. 2019, 36, 152. [Google Scholar] [CrossRef]
- Halley, J.; Chou, Y.R.; Cicchino, C.; Huang, M.; Sharma, V.; Tan, N.C.; Thakkar, S.; Zhou, L.L.; Al-Azzam, W.; Cornen, S.; et al. An Industry Perspective on Forced Degradation Studies of Biopharmaceuticals: Survey Outcome and Recommendations. J. Pharm. Sci. 2020, 109, 6–21. [Google Scholar] [CrossRef]
- Tamizi, E.; Jouyban, A. Forced degradation studies of biopharmaceuticals: Selection of stress conditions. Eur. J. Pharm. Biopharm. 2016, 98, 26–46. [Google Scholar] [CrossRef]
- Nowak, C.; Cheung, J.K.; Dellatore, S.M.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. mAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef]
- Ratcliffe, A.; Stanworth, D.R. Measurement of the binding activity of defined IgG aggregates to macrophage Fc receptors. Immunol. Lett. 1983, 7, 73–76. [Google Scholar] [CrossRef]
- Pestel, J.; Dessaint, J.P.; Joseph, M.; Bazin, H.; Capron, A. Macrophage triggering by aggregated immunoglobulins. II. Comparison of IgE and IgG aggregates or immune complexes. Clin. Exp. Immunol. 1984, 57, 404–412. [Google Scholar]
- Joubert, M.K.; Hokom, M.; Eakin, C.; Zhou, L.; Deshpande, M.; Baker, M.P.; Goletz, T.J.; Kerwin, B.A.; Chirmule, N.; Narhi, L.O.; et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 2012, 287, 25266–25279. [Google Scholar] [CrossRef]
- Telikepalli, S.; Shinogle, H.E.; Thapa, P.S.; Kim, J.H.; Deshpande, M.; Jawa, V.; Middaugh, C.R.; Narhi, L.O.; Joubert, M.K.; Volkin, D.B. Physical characterization and in vitro biological impact of highly aggregated antibodies separated into size-enriched populations by fluorescence-activated cell sorting. J. Pharm. Sci. 2015, 104, 1575–1591. [Google Scholar] [PubMed]
- Polumuri, S.K.; Haile, L.A.; Ireland, D.D.C.; Verthelyi, D. Aggregates of IVIG or Avastin, but not HSA, modify the response to model innate immune response modulating impurities. Sci. Rep. 2018, 8, 11477. [Google Scholar] [CrossRef] [PubMed]
- Moussa, E.M.; Kotarek, J.; Blum, J.S.; Marszal, E.; Topp, E.M. Physical Characterization and Innate Immunogenicity of Aggregated Intravenous Immunoglobulin (IGIV) in an In Vitro Cell-Based Model. Pharm. Res. 2016, 33, 1736–1751. [Google Scholar] [PubMed]
- Tada, M.; Aoyama, M.; Ishii-Watabe, A. Fcg Receptor Activation by Human Monoclonal Antibody Aggregates. J. Pharm. Sci. 2020, 109, 576–583. [Google Scholar]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J. Immunol. 2013, 190, 4315–4323. [Google Scholar]
- Li, P.; Jiang, N.; Nagarajan, S.; Wohlhueter, R.; Selvaraj, P.; Zhu, C. Affinity and kinetic analysis of Fcgamma receptor IIIa (CD16a) binding to IgG ligands. J. Biol. Chem. 2007, 282, 6210–6221. [Google Scholar]
- Luo, Y.; Lu, Z.; Raso, S.W.; Entrican, C.; Tangarone, B. Dimers and multimers of monoclonal IgG1 exhibit higher in vitro binding affinities to Fcgamma receptors. mAbs 2009, 1, 491–504. [Google Scholar] [CrossRef]
- Dorion-Thibaudeau, J.; Raymond, C.; Lattova, E.; Perreault, H.; Durocher, Y.; De Crescenzo, G. Towards the development of a surface plasmon resonance assay to evaluate the glycosylation pattern of monoclonal antibodies using the extracellular domains of CD16a and CD64. J. Immunol. Methods 2014, 408, 24–34. [Google Scholar]
- Lopez, E.; Scott, N.E.; Wines, B.D.; Hogarth, P.M.; Wheatley, A.K.; Kent, S.J.; Chung, A.W. Low pH Exposure During Immunoglobulin G Purification Methods Results in Aggregates That Avidly Bind Fcgamma Receptors: Implications for Measuring Fc Dependent Antibody Functions. Front. Immunol. 2019, 10, 2415. [Google Scholar]
- Geuijen, K.P.M.; Oppers-Tiemissen, C.; Egging, D.F.; Simons, P.J.; Boon, L.; Schasfoort, R.B.M.; Eppink, M.H.M. Rapid screening of IgG quality attributes—Effects on Fc receptor binding. FEBS Open Bio 2017, 7, 1557–1574. [Google Scholar]
- Bajardi-Taccioli, A.; Co, C.; Bond, C.; Masci, A.; Brantley, T.; Xu, C.; Bergelson, S.; Feschenko, M. Biolayer Interferometry-based FcgammaRIIa binding assay for a therapeutic antibody with strong effector function. Anal. Biochem. 2020, 611, 113842. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Dash, R.; Rathore, A.S. Impact of mAb Aggregation on Its Biological Activity: Rituximab as a Case Study. J. Pharm. Sci. 2020, 109, 2684–2698. [Google Scholar] [PubMed]
- Nagashima, H.; Tezuka, T.; Tsuchida, W.; Maeda, H.; Kohroki, J.; Masuho, Y. Tandemly repeated Fc domain augments binding avidities of antibodies for Fcgamma receptors, resulting in enhanced antibody-dependent cellular cytotoxicity. Mol. Immunol. 2008, 45, 2752–2763. [Google Scholar]
- Nagashima, H.; Ootsubo, M.; Fukazawa, M.; Motoi, S.; Konakahara, S.; Masuho, Y. Enhanced antibody-dependent cellular phagocytosis by chimeric monoclonal antibodies with tandemly repeated Fc domains. J. Biosci. Bioeng. 2011, 111, 391–396. [Google Scholar]
- International Conference on Harmonisation. Stability Testing: Photostability Testing of New Drug Substance and Products Q1B; ICH Harmonised Tripartite Guideline; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 1996. [Google Scholar]
- Cain, P.; Huang, L.; Tang, Y.; Anguiano, V.; Feng, Y. Impact of IgG subclass on monoclonal antibody developability. mAbs 2023, 15, 2191302. [Google Scholar]
- Shah, D.D.; Zhang, J.; Hsieh, M.C.; Sundaram, S.; Maity, H.; Mallela, K.M.G. Effect of Peroxide- Versus Alkoxyl-Induced Chemical Oxidation on the Structure, Stability, Aggregation, and Function of a Therapeutic Monoclonal Antibody. J. Pharm. Sci. 2018, 107, 2789–2803. [Google Scholar]
- Cross, R.W.; Wiethoff, C.M.; Brown-Augsburger, P.; Berens, S.; Blackbourne, J.; Liu, L.; Wu, X.; Tetreault, J.; Dodd, C.; Sina, R.; et al. The Therapeutic Monoclonal Antibody Bamlanivimab Does Not Enhance SARS-CoV-2 Infection by FcR-Mediated Mechanisms. Pathogens 2023, 12, 1408. [Google Scholar] [CrossRef]
- Gottschalk, P.G.; Dunn, J.R. Measuring parallelism, linearity, and relative potency in bioassay and immunoassay data. J. Biopharm. Stat. 2005, 15, 437–463. [Google Scholar]
- Faya, P.; Zhang, T.; Walton, W.; Novick, S. Non-constant mean relative potency for antibody-dependent cellular cytotoxicity assays. J. Biopharm. Stat. 2024, 1–12. [Google Scholar] [CrossRef]
- Parekh, B.S.; Berger, E.; Sibley, S.; Cahya, S.; Xiao, L.; LaCerte, M.A.; Vaillancourt, P.; Wooden, S.; Gately, D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. mAbs 2012, 4, 310–318. [Google Scholar]
- Qi, P.; Volkin, D.B.; Zhao, H.; Nedved, M.L.; Hughes, R.; Bass, R.; Yi, S.C.; Panek, M.E.; Wang, D.; Dalmonte, P.; et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J. Pharm. Sci. 2009, 98, 3117–3130. [Google Scholar] [PubMed]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [PubMed]
- Gao, X.; Ji, J.A.; Veeravalli, K.; Wang, Y.J.; Zhang, T.; McGreevy, W.; Zheng, K.; Kelley, R.F.; Laird, M.W.; Liu, J.; et al. Effect of individual Fc methionine oxidation on FcRn binding: Met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J. Pharm. Sci. 2015, 104, 368–377. [Google Scholar] [PubMed]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Li, Y.; Drummond, J.; Prueksaritanont, T.; et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar]
- Bommana, R.; Chai, Q.; Schoneich, C.; Weiss, W.F.; Majumdar, R. Understanding the Increased Aggregation Propensity of a Light-Exposed IgG1 Monoclonal Antibody Using Hydrogen Exchange Mass Spectrometry, Biophysical Characterization, and Structural Analysis. J. Pharm. Sci. 2018, 107, 1498–1511. [Google Scholar]
- Zheng, K.; Ren, D.; Wang, Y.J.; Lilyestrom, W.; Scherer, T.; Hong, J.K.Y.; Ji, J.A. Monoclonal Antibody Aggregation Associated with Free Radical Induced Oxidation. Int. J. Mol. Sci. 2021, 22, 3952. [Google Scholar] [CrossRef]
- Forest-Nault, C.; Gaudreault, J.; Henry, O.; Durocher, Y.; De Crescenzo, G. On the Use of Surface Plasmon Resonance Biosensing to Understand IgG-FcgammaR Interactions. Int. J. Mol. Sci. 2021, 22, 6616. [Google Scholar]
- Holtrop, T.; Budding, K.; Brandsma, A.M.; Leusen, J.H.W. Targeting the high affinity receptor, FcgammaRI, in autoimmune disease, neuropathy, and cancer. Immunother. Adv. 2022, 2, ltac011. [Google Scholar]
- Ramsland, P.A.; Farrugia, W.; Bradford, T.M.; Sardjono, C.T.; Esparon, S.; Trist, H.M.; Powell, M.S.; Tan, P.S.; Cendron, A.C.; Wines, B.D.; et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J. Immunol. 2011, 187, 3208–3217. [Google Scholar]
- Suzuki, T.; Hashii, N.; Tada, M.; Ishii-Watabe, A. The influence of antibody engineering on Fc conformation and Fc receptor binding properties: Analysis of FcRn-binding engineered antibodies and an Fc fusion protein. mAbs 2021, 13, 1923366. [Google Scholar] [CrossRef]
- Grevys, A.; Bern, M.; Foss, S.; Bratlie, D.B.; Moen, A.; Gunnarsen, K.S.; Aase, A.; Michaelsen, T.E.; Sandlie, I.; Andersen, J.T. Fc Engineering of Human IgG1 for Altered Binding to the Neonatal Fc Receptor Affects Fc Effector Functions. J. Immunol. 2015, 194, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Thomann, M.; Wegele, H.; Avenal, C.; Schlothauer, T.; Gygax, D.; Beck, H. Oxidation of M252 but not M428 in hu-IgG1 is responsible for decreased binding to and activation of hu-FcgammaRIIa (His131). Biologicals 2017, 50, 125–128. [Google Scholar] [PubMed]
- Duchemin, A.M.; Ernst, L.K.; Anderson, C.L. Clustering of the high affinity Fc receptor for immunoglobulin G (Fc gamma RI) results in phosphorylation of its associated gamma-chain. J. Biol. Chem. 1994, 269, 12111–12117. [Google Scholar] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar]
- Falconer, D.J.; Subedi, G.P.; Marcella, A.M.; Barb, A.W. Antibody Fucosylation Lowers the FcgammaRIIIa/CD16a Affinity by Limiting the Conformations Sampled by the N162-Glycan. ACS Chem. Biol. 2018, 13, 2179–2189. [Google Scholar] [CrossRef]
- Caaveiro, J.M.; Kiyoshi, M.; Tsumoto, K. Structural analysis of Fc/FcgammaR complexes: A blueprint for antibody design. Immunol. Rev. 2015, 268, 201–221. [Google Scholar]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brunker, P.; Umana, P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar]
- Klein, C.; Lammens, A.; Schafer, W.; Georges, G.; Schwaiger, M.; Mossner, E.; Hopfner, K.P.; Umana, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs 2013, 5, 22–33. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Binding Relative to Ref | % Total Agg (SEC) | % Total Agg (NR CE-SDS) | % Non-Red Agg (R CE-SDS) | % Fragments (R CE-SDS) | ||
|---|---|---|---|---|---|---|---|
| FcγRIIa 131R | FcγRIIIa 158V | FcRn | |||||
| Ref | 1.0 | 1.0 | 1.0 | 0.5 | 0.2 | 0.7 | 1.4 |
| Batch A—Light | 6.5 | 1.1 | 0.9 | 11.2 | 12.0 | 6.2 | 1.4 |
| Batch A—Dark | 1.1 | 1.0 | 1.0 | 0.6 | 0.5 | 0.7 | 1.4 |
| Batch B—Light | 2.8 | 0.9 | 0.9 | 3.7 | 4.1 | 5.1 | 1.5 |
| Batch B—Dark | 1.0 | 0.9 | 1.0 | 0.3 | 0.4 | 0.6 | 1.4 |
| Condition | FcγRIIa Binding * | % Total Agg (SEC) | % Total Agg (NR CE-SDS) | % Non-Red Agg (R CE-SDS) | % Fragments (R CE-SDS) |
|---|---|---|---|---|---|
| Ref | 1.0 | 0.5 | 0.2 | 0.7 | 1.4 |
| Temp Only | 1.2 | 0.7 | 0.6 | 1.1 | 2.5 |
| pH 4.0 | 1.2 | 0.5 | 0.5 | 0.7 | 3.7 |
| pH 8.0 | 1.8 | 1.9 | 1.4 | 5.6 | 3.1 |
| 10 ppm H2O2 | 1.2 | 0.8 | 0.7 | 1.1 | 2.5 |
| 20 ppm Fe3+ | 1.9 | 1.6 | 1.1 | 1.4 | 2.9 |
| 20 ppm Cu2+ | 1.4 | 1.0 | 0.9 | 6.2 | 2.5 |
| 200 mM Glucose | 1.3 | 1.1 | 0.9 | 1.6 | 16.8 |
| 1 ppm H2O2 | 1.2 | 0.7 | 0.7 | 1.1 | 2.6 |
| 0.5 ppm Fe3+ | 2.0 | 2.4 | 1.6 | 2.1 | 3.0 |
| Sample | SEC | % Total Agg (NR CE-SDS) | % Non-Red Agg (R CE-SDS) | % Fragments (R CE-SDS) | |||
|---|---|---|---|---|---|---|---|
| % Monomer | % Dimer | % H.O. Agg | % Total Agg | ||||
| Ref | 99.5 | 0.4 | 0.1 | 0.5 | 0.2 | 0.7 | 1.4 |
| 110% ICH | 89.1 | 10.1 | 0.9 | 11 | 9.1 | 5.2 | 2.3 |
| Peak 1 | 20.9 | 56.0 | 23.0 | 79.0 | 72.9 | 21 | 2.6 |
| Peak 2 | 25.2 | 70.3 | 4.5 | 74.8 | 68.7 | 19.6 | 2.4 |
| Peak 3 | 86.5 | 13.1 | 0.3 | 13.4 | 13.7 | 6.8 | 1.8 |
| Peak 4 | 97.9 | 1.99 | 0.04 | 2.0 | 2.4 | 3.4 | 2.4 |
| Sample | Binding (Relative to Ref) | ||
|---|---|---|---|
| FcγRIIa Binding | FcγRIIIa Binding | Antigen Binding | |
| Ref | 1.0 | 1.0 | 1.0 |
| 110% ICH | 4.3 | 1.8 | 1.1 |
| Peak 1 | 26.7 | 7.6 | 0.8 |
| Peak 2 | 28.6 | 6.6 | 0.8 |
| Peak 3 | 6.2 | 2.0 | 1.0 |
| Peak 4 | 1.4 | 1.0 | 0.9 |
| Sample | %M252 Oxidation | %M358 Oxidation | %M428 Oxidation | %N325 Deamidation | % %Afucosylated |
|---|---|---|---|---|---|
| Ref | 1.84 | 0.50 | 0.73 | <1 | 6.1 |
| 110% ICH | 24.45 | 4.26 | 17.82 | <1 | 5.4 |
| Peak 1 | 25.38 | 4.68 | 18.28 | <1 | 5.1 |
| Peak 2 | 25.81 | 4.37 | 17.94 | <1 | 5.3 |
| Peak 3 | 25.05 | 4.04 | 17.59 | <1 | 4.9 |
| Peak 4 | 23.85 | 4.12 | 17.09 | <1 | 5.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, W.J.; Zhang, S.J.; Wilson, J.J.; Harvey, B.N.; Clemens, M.; Gu, Y. Impact of Monoclonal Antibody Aggregates on Effector Function Characterization. Antibodies 2025, 14, 31. https://doi.org/10.3390/antib14020031
Walton WJ, Zhang SJ, Wilson JJ, Harvey BN, Clemens M, Gu Y. Impact of Monoclonal Antibody Aggregates on Effector Function Characterization. Antibodies. 2025; 14(2):31. https://doi.org/10.3390/antib14020031
Chicago/Turabian StyleWalton, Wendy J., Shousong Jason Zhang, Joseph J. Wilson, Briana N. Harvey, Matthew Clemens, and Yingmei Gu. 2025. "Impact of Monoclonal Antibody Aggregates on Effector Function Characterization" Antibodies 14, no. 2: 31. https://doi.org/10.3390/antib14020031
APA StyleWalton, W. J., Zhang, S. J., Wilson, J. J., Harvey, B. N., Clemens, M., & Gu, Y. (2025). Impact of Monoclonal Antibody Aggregates on Effector Function Characterization. Antibodies, 14(2), 31. https://doi.org/10.3390/antib14020031